NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B

1
Key Laboratory of Industrial Ecology and Environmental Engineering, Ministry of Education, School of Environmental Science and Technology, Dalian University of Technology, Dalian 116024, China
2
Shandong Hualu-Hengsheng Chemical Co., Ltd., Dezhou 253000, China
3
Department of Chemical and Engineering, Yingkou Institute of Technology, YingKou 115014, China
*
Author to whom correspondence should be addressed.
Water 2019, 11(2), 384; https://doi.org/10.3390/w11020384
Submission received: 26 January 2019
/
Revised: 19 February 2019
/
Accepted: 20 February 2019
/
Published: 22 February 2019
(This article belongs to the Special Issue Advanced Oxidation Technologies in Industrial Wastewater Treatment)
Abstract
:Magnetic spinel ferrites that act as heterogeneous catalysts and generate powerful radicals from peroxymono-sulfate (PMS) for the degradation of organic pollutants have received much attention in recent years due to the characteristic of environmental benefits. In this study, NiO-NiFe2O4-rGO magnetic nanomaterials were synthesized using a calcinated Ni-Fe-LDH-rGO precursor. The morphology, structure, and chemical constitution were characterized using X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray energy dispersive spectroscopy (EDS), transmission electron microscope (TEM), N2 adsorption-desorption isotherms, X-ray photoelectron spectroscopy (XPS), and vibrating sample magnetometer (VSM). The catalytic performance of NiO-NiFe2O4-rGO nanoparticles was thoroughly evaluated for peroxymonosulfate (PMS) activation and its removal of rhodamine B (RhB) from water. The influence of different process parameters on the RhB degradation efficiency was examined. Further, the catalytic stability was evaluated. Under optimized conditions, the NiO-NiFe2O4-rGO/PMS system was very efficient; RhB fully degraded after 40 min at room temperature. Quenching experiments and electronic paramagnetic resonance (EPR) results suggested that SO4−· and OH· were the main active species in the degradation process. Moreover, NiO-NiFe2O4-rGO catalyst was stable without any apparent activity loss after three cycling runs.
1. Introduction
Currently, in the industrial production processes, sustainable development is a major issue. The most contaminated textile wastewater stream, dyeing discharge, was selected for dedicated treatment using advanced oxidation processes (AOPs) [1]. The basic principle of the AOPs is to generate highly active intermediate species (i.e. OH·, O2−·, and SO4−·) for mineralization of refractory organic pollutants, water pathogens, and disinfection by-products. Due to the high oxidative potential of hydroxyl radicals (OH·), which appear in AOPs, many organic compounds, including dyes, can be decomposed; thus, OH· have high second-order reaction rate constants with most organic molecules, including both bulk organic matter and micropollutants [2]. The bulk organic matter is abundantly present in secondary effluent and acts as an important scavenger for OH·, which has become much less available for reaction with organic micropollutants [3,4]. Therefore, the interest in sulfate radical (SO4−·) based AOPs has recently grown. Because of the higher selectivity of SO4−·, which reacts mainly through an electron transfer mechanism, the scavenging rate of SO4−· in bulk organic matter is 35–75 times lower when compared to that of OH· [5,6]. Other advantages of SO4−· are the high oxidation reduction potential (2.5–3.1 v) and the potential to oxidize and degrade refractory organic pollutants in wastewater. At the same time, it has a long half-life and a wide usage range of pH [7]. Therefore, it has more potential in the field of pollutant removal.
Usually, sulfate radicals can be obtained through persulfate (PMS) and sulfates activation via heat, light, radiation, or chemical activation, with which the oxidative properties can be significantly enhanced to form the free radicals [8], while PMS and sulfates have certain oxidation properties without activation. Therefore, many scholars have paid attention to the activation of PMS and sulfates, as well as the production of more sulfate radicals. Spinel oxides are widely used in catalysis, energy storage, environmental restoration, and other fields, due to its good catalytic activity and magnetic characteristics [9]. For example, Boukherroub et al. demonstrated that in less than 10 min at room temperature a spinel Co2SnO4/PMS system proved very efficient with a full degradation of rhodamine B (RhB) and pentachlorophenol (PCP) [10]. Ren et al. evaluated the performance of CoFe2O4, CuFe2O4, MnFe2O4, and ZnFe2O4 in PMS oxidation and concluded that CoFe2O4 possessed superior catalytic activity towards PMS for the degradation of dibutyl phthalate and that all catalysts presented favorable recycling and stability in the repeated batch experiment [11]. However, spinel ferrite is easy to reunite in the synthesis process and its catalytic properties are affected significantly [12]. The effect could be overcome while composited with reductive graphene (rGO) and a high conductivity and high specific surface area. Yao et al. and Xu et al. deposited MnFe2O4 and CoFe2O4 nanoparticles on reduced graphene oxide (rGO), respectively, and found that both PMS activation and organic pollutants were degraded by the obtained hybrid catalysts and could be substantially enhanced [13,14].
To the best of our knowledge, there has been no prior investigation of NiO-NiFe2O4-rGO for PMS activation. In this study, we focused on the synthesis of NiO-NiFe2O4-rGO and its magnetic nanomaterials using a hydrothermal route. Futher, we considered their ability to activate PMS for efficient degradation of RhB at room temperature. The as-prepared hybrid catalyst was characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), EDS, transmission electron microscope (TEM), N2 adsorption-desorption isotherms, X-ray photoelectron spectroscopy (XPS), and vibrating sample magnetometer (VSM). Its catalytic activity in PMS solution was evaluated using the oxidation of a xanthene zwitterionic dye rhodamine B (RhB). Moreover, we investigated the influence of various parameters related to the operational procedure. Finally, we studied the degradation mechanism that was tentatively proposed and the recyclability of magnetic hybrid catalyst.
2. Materials and Methods
2.1. Preparation of Catalyst
2.1.1. Preparation of graphene oxide(GO)
Graphene oxide (GO) was prepared via the improved Hummers method [15].
2.1.2. Preparation of Ni-Fe-LDH-rGO
Ni-Fe-LDH-rGO was prepared using the single-step hydrothermal method. The specific preparation process was as follows: Ni(NO3)2·6H2O and Fe(NO3)3·9H2O (the molar ratio was 3:1) was weighted and a certain amount of carbamide was dissolved in GO aqueous solution, which contained 300 mg GO. Then, after the deionized water was added and 160 mL solution was formed, the mixed solution was shifted into high pressure hydrothermal reaction kettle for 14 h at 413 K during magnetic stirring for 20 min at room temperature. After being cooled to the room temperature, the product was washed several times with deionized water and absolute ethyl alcohol and the Ni-Fe-LDH-rGO was acquired after being dried in vacuum drying oven for 8 h at 333 K.
2.1.3. Preparation of NiO-NiFe2O4-rGO
After calcinating for 4 h at air atmosphere (up to 773 K with the heating rate 2 K·min−1), a suitable amount of Ni-Fe-LDH-rGO was placed in a crucible and put into a muffle furnace. Then the NiO-NiFe2O4-rGO nanocomposites was obtained after being cooled to room temperature.
2.2. Characterization of Catalyst
The crystal structures of the catalysts were characterized with a powder X-ray diffraction (XRD) using Cu Kα (1.5406 Å) radiation at a scanning speed of 4°/min that ranged from 10° to 80°. The morphology of NiO-NiFe2O4-rGO was characterized using a scanning electron microscopy (SEM) (JEOL Model JSM-5600LV), equipped with an X-ray energy dispersive spectroscopy (EDS), and a FEI Tecnai G2F30 transmission electron microscope (TEM) instrument. Brunauer–Emmett–Teller (BET) surface areas, mesoporous surface areas, and micropore volumes of the catalysts were measured using nitrogen adsorption and desorption at 77.3 K with a Quantachrome Instruments NOVA 4200e apparatus. Valence states were analyzed using X-ray phtoelectron spectroscopy (XPS) (PHI 5000 Versa Probe/Scanning ESCA Microprobe, ULVAC-PHI, Inc., Chigasaki, Japan) with a monochromatized Mg K𝛼 (1253.6 eV) X-ray source. Electronic paramagnetic resonance (EPR) spectrometer (Bruker A200 spectrometer, Bruker Corporation, Berlin, Germany) with a 300 W high pressure mercury lamp was applied to obtain the information of the active species during the oxidation process. A vibrating sample magnetometer MPMS-XL-7 (Quantum Design, San Diego, CA, USA) was used to measure the magnetic properties of NiO-NiFe2O4-rGO magnetic nanomaterials.
2.3. Catalytic Test Procedures and Analytical Method
The catalytic reaction procedure was carried out in a 1.0 L beaker using rhodamine B (RhB) as the targeted contaminant. Further, 500 mL RhB solution (C0 = 20.16 mg·L−1, pH = 7.0), a certain amount of NiO-NiFe2O4-rGO catalyst, and PMS were added into the beaker, then stirred to ensure the catalysts were being fully exposed to the solution. Next, a 5 ml reaction suspension was withdrawn and filtered at regular intervals. The supernate was then analyzed using a UV-Vis spectrophotometer (Shimadzu UV1700, Japan) at 555 nm to calculate the removal efficiency of RhB [10]. To verify the mineralization of RhB, the total organic carbon (TOC) removal rate during the oxidation process was monitored by a total organic carbon analyzer (Shimadzu TOC V-CPN, Japan). All the experiments were performed in triplicates and the results of the analysis were presented as the mean with a standard deviation less than 5%.
The identification of transformation products were detected using a liquid chromatography-mass spectrometry (LC-MS, WATERS, UPLC-MS SQD2) in electrospray positive ion mode (ESI+), which was equipped with an Agilent TC-C18, 250 × 4.6 mm column. The mobile phase consisted of a mixture of methanol (30%) and ammonium acetate of 1g·L−1 in H2O (70%) at a flow rate of 0.2 mL·min−1. The detection was performed at 270 nm. At given intervals, the RhB solution was filtered through a Minisart RC filter (pore size 0.22 mm) to remove the catalyst. In addition, it was analyzed by the HPLC column. ESI source conditions were as follows: cone voltage of 3 V, capillary voltage of 3 kV, and sheath gas flow rate of 800 L·h−1. The full scan mass ranged from 0 to 1000.
3. Results and Discussion
3.1. Catalyst Characterization
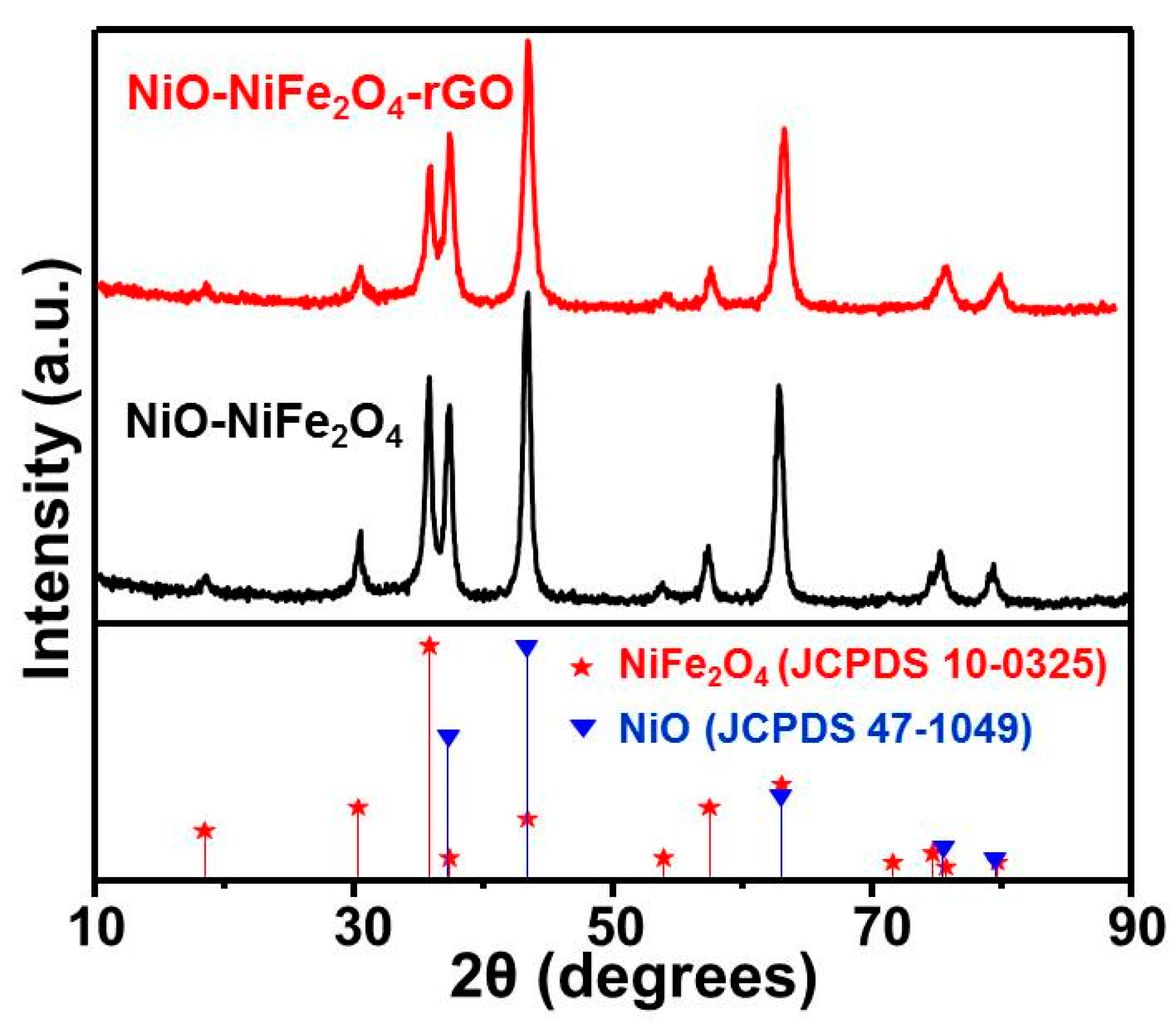
Figure 1 is an XRD pattern of NiO-NiFe2O4 and NiO-NiFe2O4-rGO. For comparison, the standard card of NiO (JCPDS 47-1049) and NiFe2O4 (JCPDS 10-0325) were given. As shown in Figure 1, the diffraction peaks were located at ca. 18.18°, 30.15°, 35.55°, 37.02°, 43.14°, 53.69°, 57.32, 62.84°, 75.37°, and 79.49°, which correspond to (111), (220), (311), (222), (400), (422), (511), (440), (622), and (444) lattice plane of NiFe2O4(JCPDS 10-0325), respectively. Further, the diffraction peaks were at 37.25°, 43.28°, 62.88°, 75.42°, and 79.41° and were ascribed to (111), (200), (220), (311), and (222) lattice plane of NiO (JCPDS 47-1049). As for NiO-NiFe2O4, the characteristic diffraction peaks were observed. The results above indicated that NiO and NiFe2O4 coexisted on a NiO-NiFe2O4 magnetic nanomaterial and the product obtained using a calcinated Ni-Fe-LDH precursor at this temperature was a nanocomposite of NiO and NiFe2O4. The XRD pattern of NiO-NiFe2O4-rGO was the same as that of NiO-NiFe2O4 and no other diffraction peaks appeared, which was mainly due to the effective prevention of rGO accumulation, leading to the reduction of the crystallization degree and the characteristic peak.
The SEM images in Figure 2a,b revealed that the NiO-NiFe2O4-rGO nanocomposite presented a laminar structure composed of discrete slices. EDS element analysis in Figure 2c–f showed that the NiO-NiFe2O4-rGO nanocomposite had a relative homogeneous distribution of C, O, Ni, and Fe elements; no other contaminated elements was detected. The element compositions of Ni and Fe using EDS analysis were confirmed with a Ni and Fe wt% of 1.27% and 3.44%, respectively, which correlated well with the nominal loading molar ratio of Ni/Fe in the starting salt solution.
TEM and HRTEM images of NiO-NiFe2O4-rGO are presented in Figure 3. As shown in Figure 3a, the distribution of NiO-NiFe2O4-rGO particle was relatively uniform. Interplanar spacing of 0.296 nm and 0.487 nm corresponded with (220) and (111) lattice plane of NiFe2O4, respectively. Further, 0.242 nm was ascribed to the (111) lattice plane of NiO, indicating that NiO and NiFe2O4 were coexisted in the NiO-NiFe2O4-rGO, which was highly dispersed at the nanometer scale.
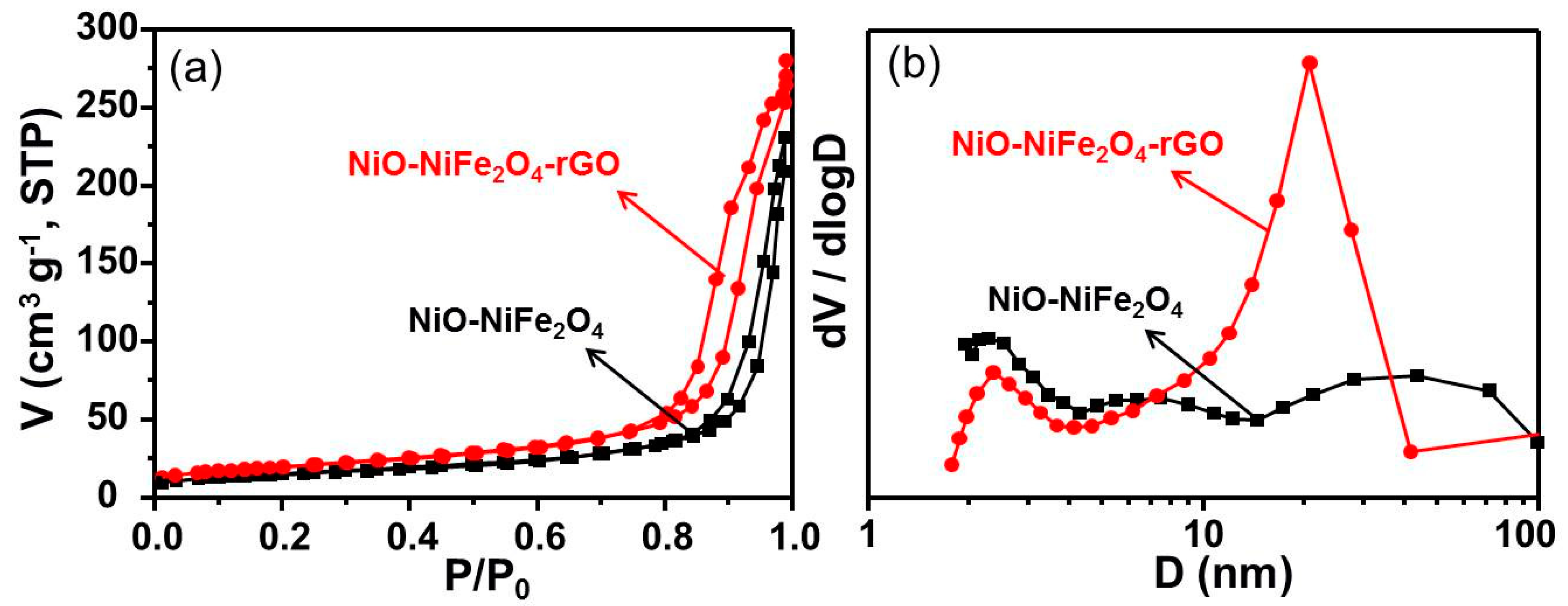
As shown in Figure 4a, an IV type isotherm was displayed with a H3 type hysteresis loop, which implied the mesoporous structure. The NiO-NiFe2O4 showed a broad hysteresis loop in a relative pressure (P/P0) range of 0.90–1.00, while the hysteresis loop of NiO-NiFe2O4-rGO became wider from 0.90 to 0.80, indicating that it had a more porous structure. In Figure 4b, it shows how the single-modal pore size distribution of NiO-NiFe2O4-rGO centered at ca. 11 nm. In Table 1, it shows the BET surface area’s total pore volume and average pore size. A higher BET specific surface area (70.6 vs. 56.29 m2·g−1) and a slightly larger pore volume (0.43 vs. 0.36 cm3·g−1) of NiO-NiFe2O4-rGO than NiO-NiFe2O4 were observed, corresponding well with the hysteresis loop features. Additionally, the introduction of rGO resulted in a more concentrated pore size distribution and a more abundant specific surface area and pore volume.
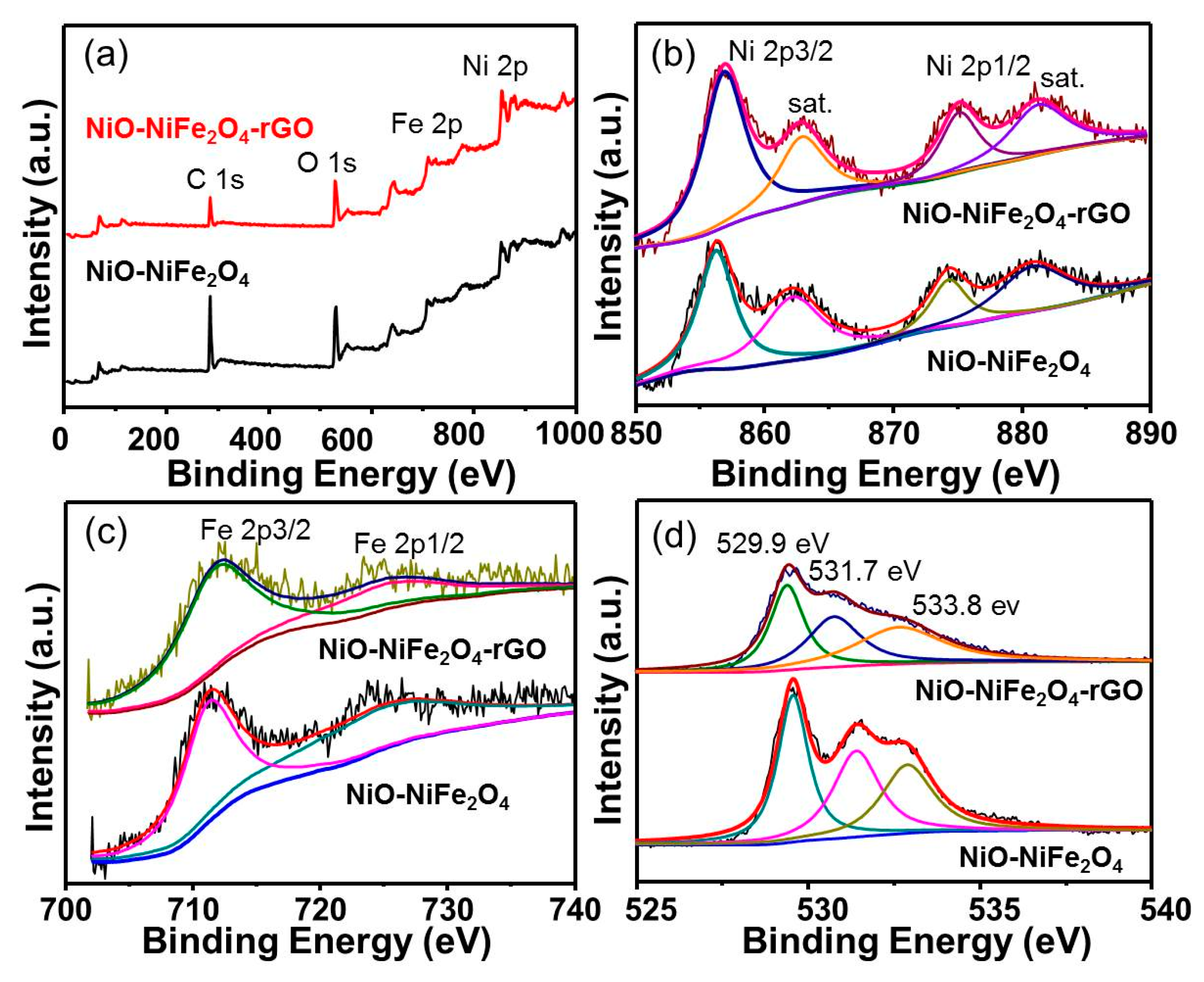
To further investigate the surface composition and valence state, XPS was also carried out on NiO-NiFe2O4 and NiO-NiFe2O4-rGO. As shown in Figure 5a, obvious O1s, C1s, and relatively weak Ni2p and Fe3d peaks were found within the observed region. The XPS spectrum of the Ni2p3/2 region could be deconvoluted into two peaks corresponding to the binding energies of approximately 855.3 and 862.4 eV, while t he Ni2p1/2 region could be deconvoluted into two peaks corresponding to the binding energies of approximately 872.9 and 880.4 eV (Figure 5b), indicating that the element Ni was existed in NiO-NiFe2O4 and NiO-NiFe2O4-rGO in the form of Ni2+ [16]. Two characteristic peaks in Figure 5c were centered at ca. 711.6 and 724.9 eV due to Fe2p3/2 and Fe2p1/2, respectively, which were associated with the Fe3+ species [17]. The corresponding O1s spectrum of NiO-NiFe2O4-rGO in Figure 5d could be decomposed into three peaks: 529.9 eV, 531.7 eV, and 533.8 eV, corresponding to the oxygen defect states and the adsorbed hydroxyl [18], respectively.
3.2. Activation of PMS by NiO-NiFe2O4-rGO for RhB Degradation
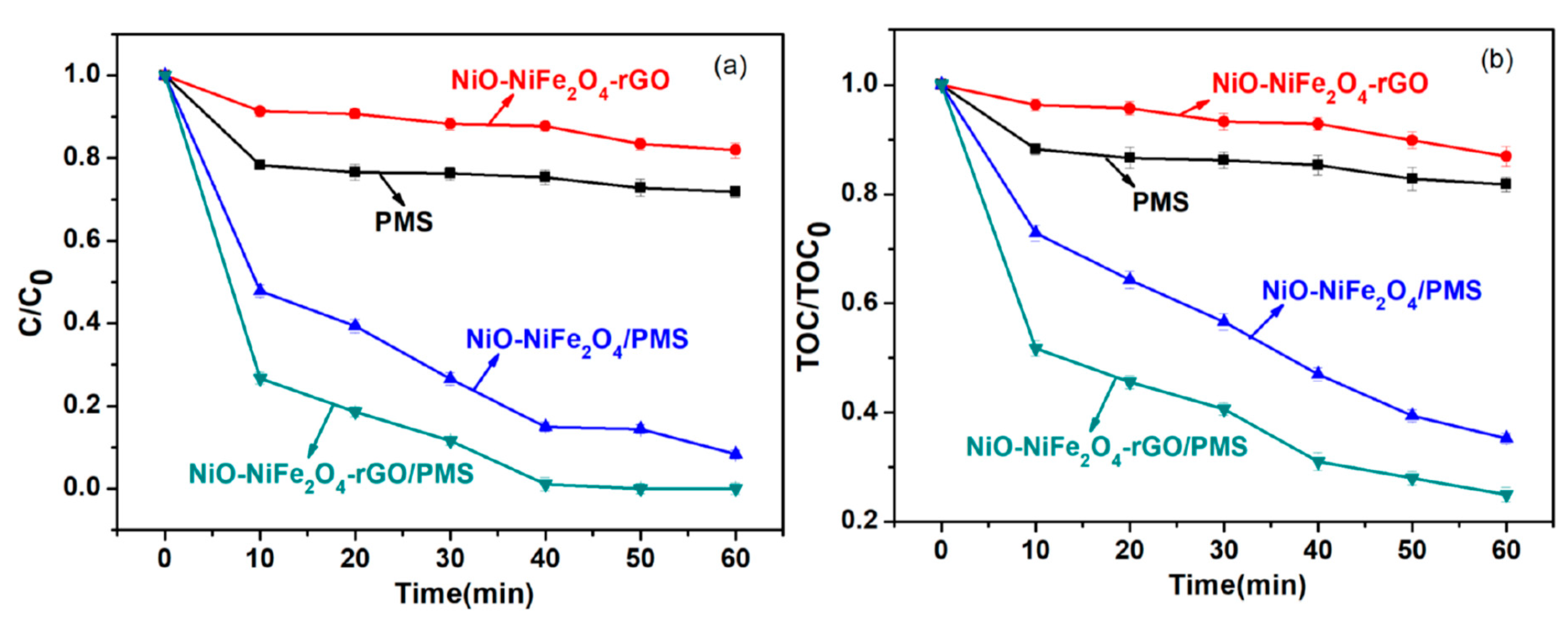
To evaluate the activation of PMS via NiO-NiFe2O4-rGO, the degradation of RhB was selected as a model reaction. For comparison, we also investigate the degradation of RhB via PMS, NiO-NiFe2O4-rGO, and NiO-NiFe2O4 alone activated PMS (shown in Figure 6a). For the individual PMS system, the removal rate of RhB was only 21% after 1 h, the reason being that PMS was relatively stable under normal temperature in spite of its high oxidization. For the individual NiO-NiFe2O4-rGO system, 12% of RhB was removed due to the adsorption. The NiO-NiFe2O4-rGO/PMS system exhibited a better removal effect and nearly 100% of RhB was removed after 40 min, while for the NiO-NiFe2O4/PMS system, only 83.6% RhB was removed in the same condition. The main reason might be that NiO-NiFe2O4-rGO and NiO-NiFe2O4 could activate PMS to form OH· and SO4−·, which had a stronger oxidizing capacity [19], as shown in the Equations (1)–(3).
To better understand the degradation of RhB in systems, the mineralization of RhB was studied with the TOC measurement method, which was shown in Figure 6b. Within 60 min, the TOC was removed by 11.2% and 18.5%, respectively, in NiO-NiFe2O4-rGO and PMS systems. At the same time, the TOC removal rate was observed to be up to 63.5% and 90.0%, respectively, in NiO-NiFe2O4/PMS and NiO-NiFe2O4-rGO/PMS systems at identical conditions. It could be illustrated that RhB had been completely degraded into the NiO-NiFe2O4-rGO/PMS system, while only part of it was subjected to the complete decomposition of CO2 and H2O.
3.3. The Effect of Different Process Parameters on the RhB Degradation
3.3.1. The Effect of NiO-NiFe2O4-rGO Dosage on RhB Degradation
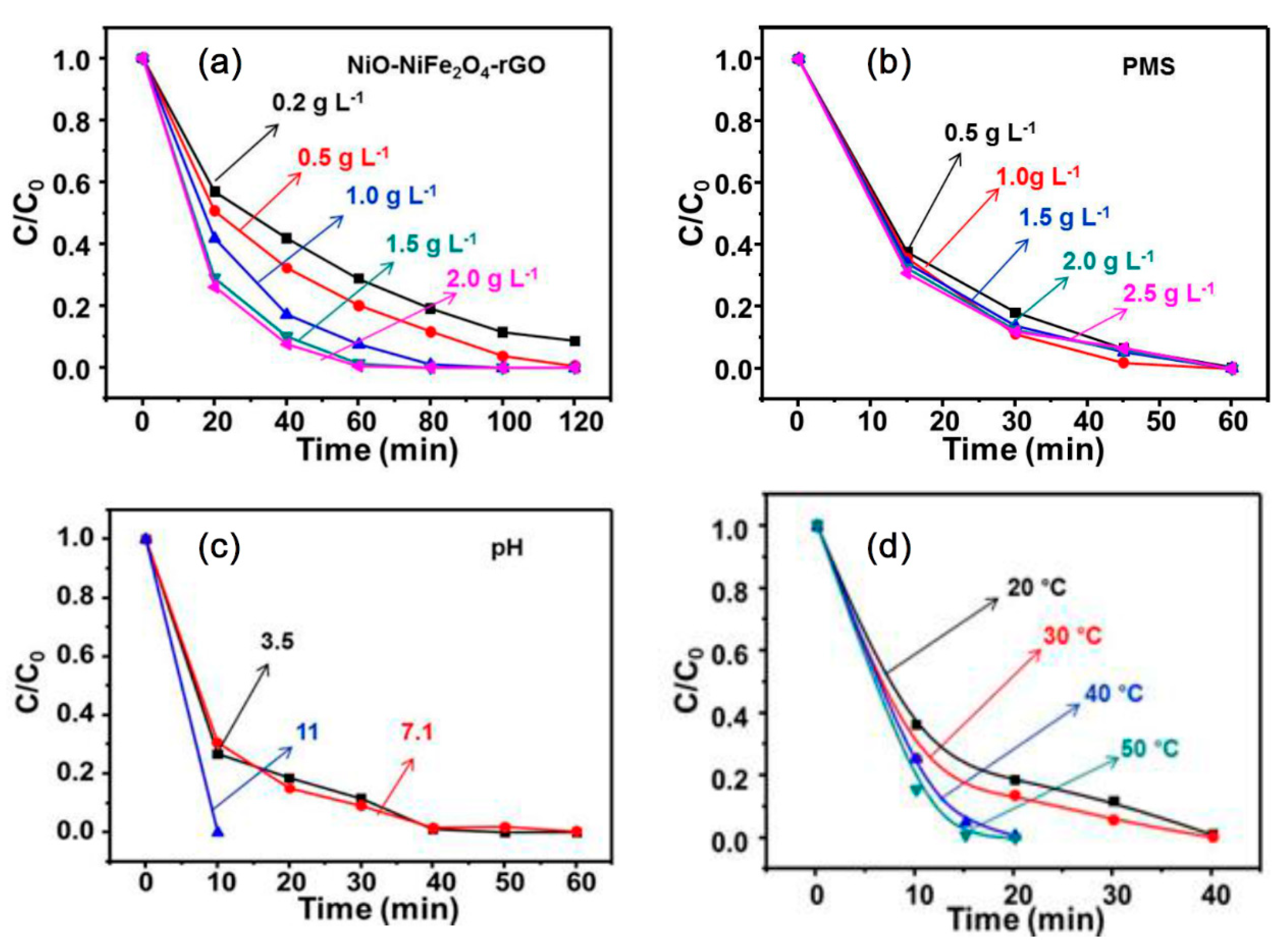
We investigated the removal effect of RhB in the range of 0.2–2.0 g·L−1 of NiO-NiFe2O4-rGO at the conditions of 20.16 mg·L−1 RhB, initial pH of 7.00, PMS of 1 g·L−1, and the temperature of 393 K. Figure 7a showed the influence of NiO-NiFe2O4-rGO dosage on RhB degradation using PMS activation. With the increase of NiO-NiFe2O4-rGO, a faster RhB degradation was observed. This is because the wider NiO-NiFe2O4-rGO surface area increased the number of active sites for adsorption and PMS activation. With a further increase of NiO-NiFe2O4-rGO dosage, more than 1.5 g·L−1, the effect was not obvious. Considering the contribution made by NiO-NiFe2O4-rGO, 1.5 g·L−1 was the most efficient dosage in the NiO-NiFe2O4-rGO /PMS combined system under this condition.
3.3.2. Effect of PMS Dosage on RhB Degradation
Figure 7b shows the impact of initial PMS dosage on the removal of RhB at an initial pH of 7.00. The increase of PMS concentration could accelerate RhB degradation. The removal rate of RhB increased from 80.0% to 90.7% with the PMS dosage increasing from 0.5 to 1.0 g·L−1 in 30 min, respectively, and then kept almost unchanged with an initial PMS dosage further increasing to 2.5 g·L−1 at the catalyst dosage of 1.5 g·L−1. It was reported that SO4−· produced using the PMS activation reacted with superfluous PMS to form SO42− [20], as shown in reaction Equation (4). Therefore, 1.0 g·L−1 was the most efficient PMS dosage.
3.3.3. Effect of Initial pH on RhB Degradation
The influence of the initial pH was studied in a NiO-NiFe2O4-rGO dosage of 1.5 g·L−1 and a PMS dosage of 1.0 g·L−1. The pH of aqueous played a significant role in RhB degradation, as shown in Figure 7c. The following order in RhB degradation in the NiO-NiFe2O4-rGO/PMS combined system at a different initial pH was observed: (initial pH 11) > (initial pH 7.1) > (initial pH 3.1). As the solution pH was in the 3–9 range, OH−/H2O was oxidized into OH· by SO4−·, and/or HSO5− was positioned at a moderate rate (Equations (2) and (3)). A relatively high degradation efficiency was maintained by the presence of both SO4−· (dominator) and OH·. However, as the solution pH was increased to more than 10, OH· scavenged SO4−·, becoming the dominator, and the oxidation capacity became weaker than SO4−· (shown in the reaction Equation (5)) [21]. After the PMS was added, the initial pH value of RhB solution was decreased [22]. With the addition of PMS, the initial pH value was decreased from 11.0 to 8.3. Actually, the reaction was under the pH of 8.3. Therefore, the fastest removal rate of RhB occurred at an initial pH of 11.
3.3.4. The Effect of the Temperature on RhB Degradation
Figure 7d illustrates how the removal rate of RhB was accelerated with the increase of temperature. Temperature, of course, produces more heat energy, thus overcoming the excitation and making the peroxygen bond rupture produce more SO4−·. This process was similar to the thermal activation, as shown in the reaction Equation (6). On the other hand, the increase of temperature could increase the collision rate between molecules and speed up the reaction to promote the degradation reaction [23].
Table 2 gave the reaction rate at different reaction temperature. Based on the Arrhenius equation dlnk/dT = Ea/RT2, activation energy of the catalytic system was calculated as 29.1 kJ·mol−1, which was relatively small. Therefore, NiO-NiFe2O4-rGO had a high catalytic activity.
3.4. Effect of Common Anions on RhB Degradation
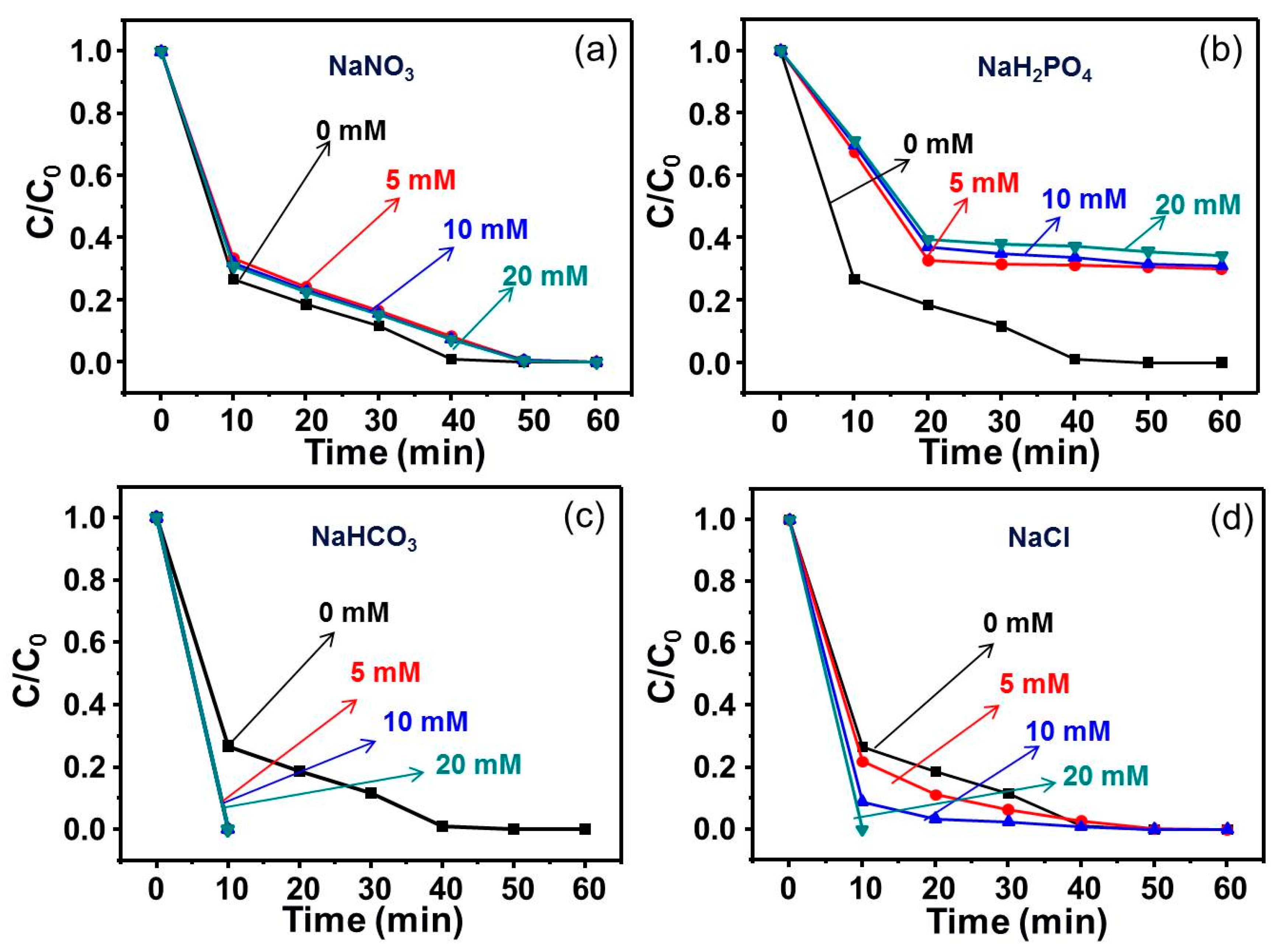
Figure 8 presents, the degradation of RhB via PMS under anion conditions ((a) NO3−, (b) H2PO4−, (c) HCO3−, and (d) Cl−). As indicated in Figure 8a, the influence of NO3− on the degradation of RhB was less. From Figure 8b, it was found that HPO42− could inhibit PMS to degrade RhB, which did not change with the HPO42− anion dosage. As reported in the literature [24], phosphate ions demonstrated a scavenging effect for sulfate radical as well as hydroxyl radical. Moreover, phosphate ions have a more apparently abominable effect on the catalysts through chelating reactions, which decreases the active species on the catalyst surface. As shown in Figure 8c, HCO3− could accelerate the degradation of RhB. It was speculated that: (1) HCO3− was capable of activating PMS to generate active species for the degradation of RhB [22] and (2) the presence of highly concentrated HCO3− could maintain the degradation system in an alkaline condition, and PMS could be decomposed under alkaline conditions, thus the degradation effect would be greatly improved [25]. As shown in Figure 8d, Cl− activated PMS to degrade RhB and the degradation of RhB became faster with the increasing concentration of Cl− anions. The reason was that in addition to SO4−·, the system would produce some hypochlorite oxidants, which also contributed to the degradation system [26].
3.5. Mechanism of PMS Activation in NiO-NiFe2O4-rGO/PMS System
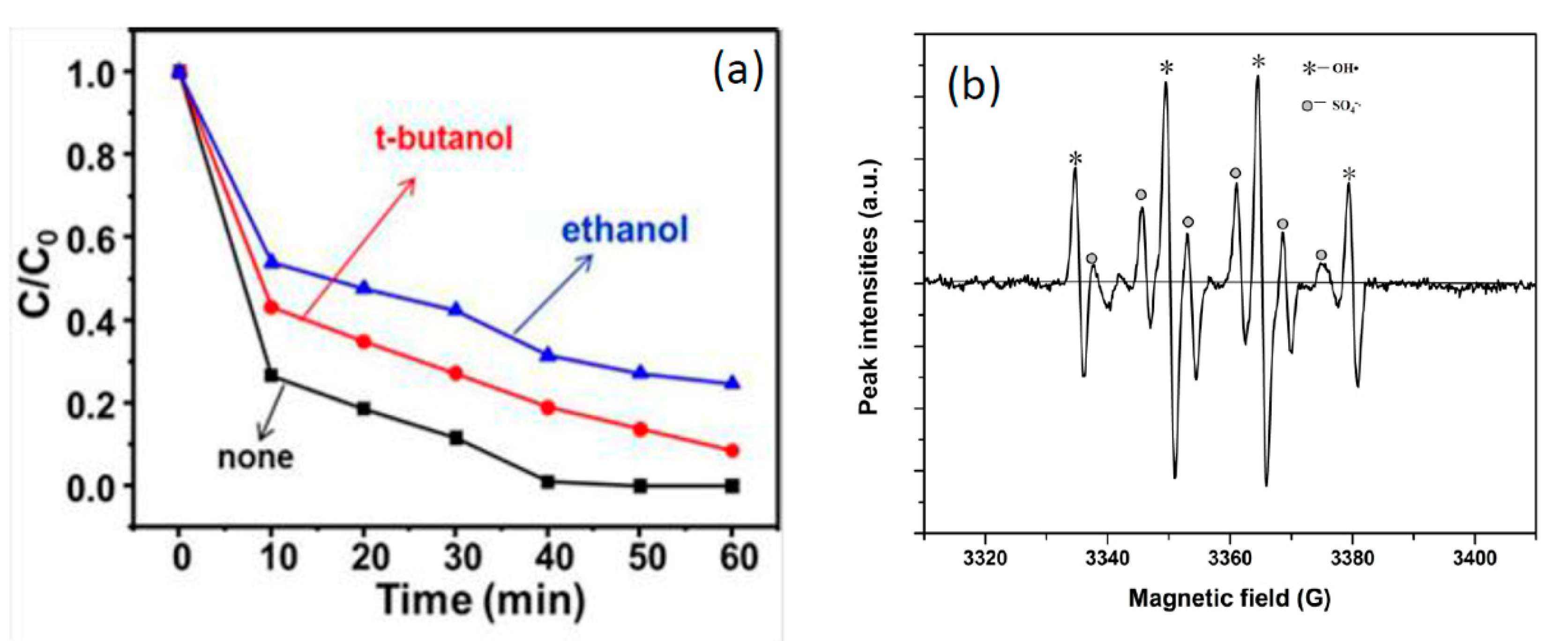
It had been reported that SO4−· was produced as PMS was activated and that SO4−· could further react with H2O or OH− to form OH·. Both SO4−· and OH· were responsible for the contaminant degradation. Tertiary butanol (t-BuOH) was the trapping agent of HO·, while ethyl alcohol(EtOH), containing α-H could be used as the trapping agent of SO4−· and HO· [27]. According to different capture rates, the purpose of identifying free radical species could be achieved. Figure 9a presents the effect of t-BuOH and EtOH on the degradation of RhB. An almost complete removal of RhB at 40 min was obtained with the absence of t-BuOH and EtOH. However, there was a 20% and 33% drop in the degradation efficiency in the presence of t-BuOH at 2 mL and EtOH at 2 mL, respectively. The results showed that SO4−· and HO· were the main active free radicals in the reaction system.
SO4−· and HO· were the most likely active free radicals in the PMS system. Therefore, the free radicals generated in the NiO-NiFe2O4-rGO/PMS system were further identified with EPR using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the spin-trapping agent. As shown in Figure 9b, DMPO-SO4 (six lines, 1:1:1:1:1:1) and DMPO-OH (four lines, 1:2:2:1) signals could be attributed to the formation of SO4−· and OH·, respectively. These results indicated that the significant amounts of SO4−· and OH· were generated during PMS activation by NiO-NiFe2O4-rGO.
3.6. Degradation Pathway of RhB in NiO-NiFe2O4-rGO/PMS System
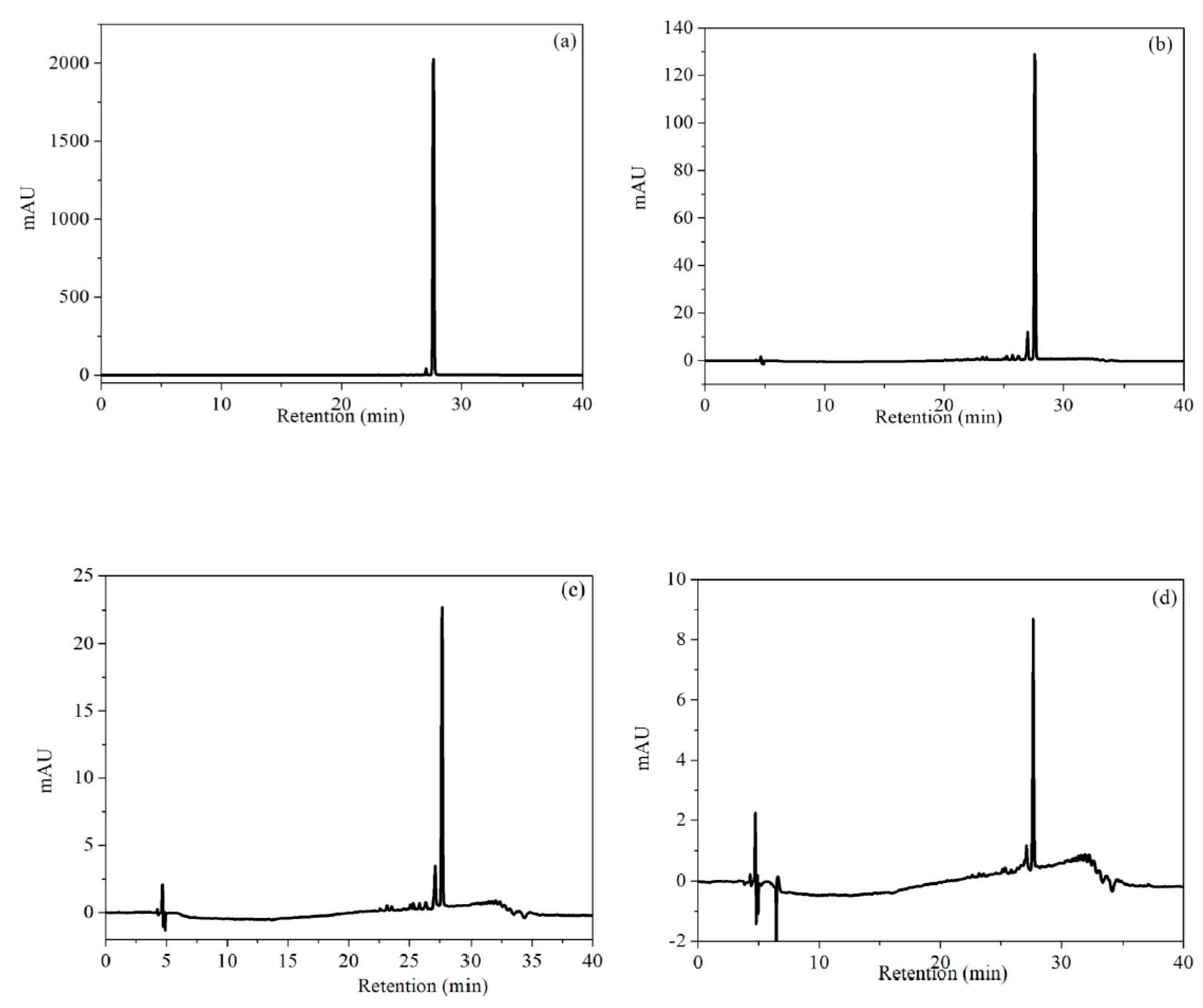
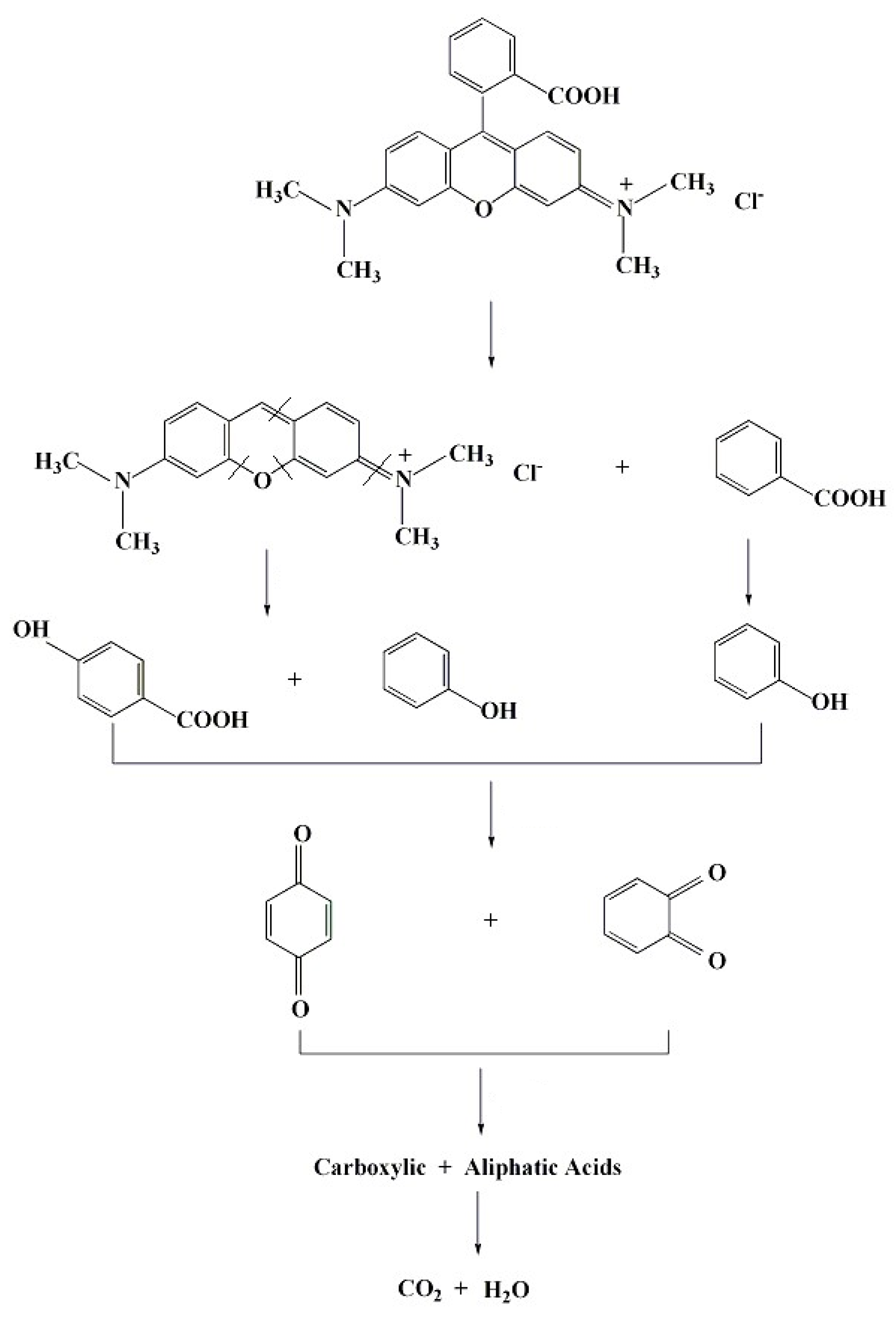
To gain further insights into the degradation process and intermediates, the reaction mixture was monitored using LC-MS equipped with a UV-vis detector at 270 nm. The chromatograms of RhB were recorded after its reaction with NiO-NiFe2O4-rGO in the presence of PMS for 0, 10, 20, and 40 min d (Figure 10). The RhB peak with a retention time of 27.6 min, under the experimental conditions, completely disappeared after 40 min. The result suggested that RhB degradation was fast, with full destruction of the conjugated structure. After a reaction of 10 min, a new peak with a retention time of 27.06 min and a charge-to-mass ratio of 416 appeared, indicating that it was a quasi-molecular ion peak of RhB molecule that lost ethyl [28]. With the prolonged reaction time, new peaks corresponded to the reaction intermediates, which was due to the Benzene ring fracture of an RhB molecule appearing at the retention time of 22–26 min. Due to the easier further degradation and the lower concentration, it was unidentified by MS. According to the experimental results above, the degradation pathway of RhB was as follows (shown in Figure 11): benzene amino and carbonyl bond of color base group were attacked to form colorless organic intermediates such as phenol, p-hydroxy benzoic acid, and further degraded to generate carboxylic acid, fatty acid, then finally completely mineralized to CO2 and H2O. In addition, in the degradation process of RhB, deethylation reaction happened. Some RhB molecules lost an ethy, and then were attacked to realize complete degradation.
3.7. Recyclability of NiO-NiFe2O4-rGO to Activate PMS
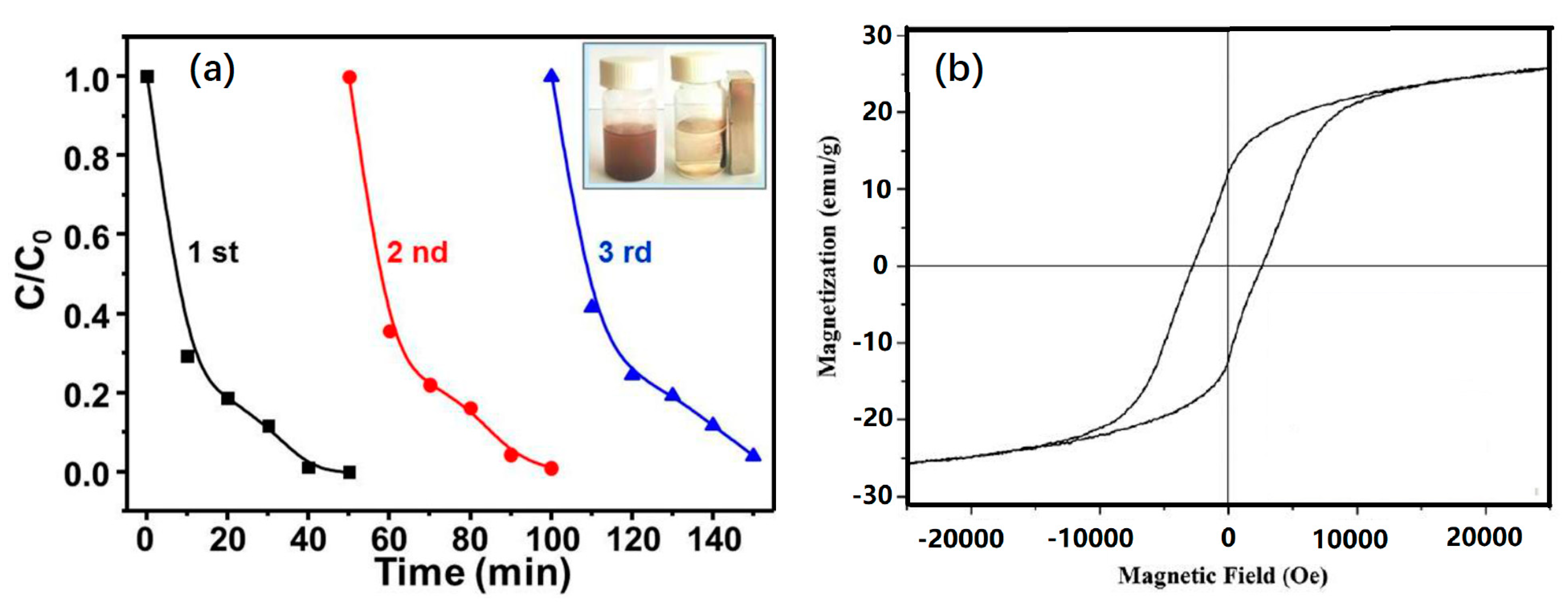
Although it was shown that the PMS was activated using NiO-NiFe2O4-rGO to yield sulfate radicals, it was necessary to investigate whether NiO-NiFe2O4-rGO could be reused as a heterogeneous catalyst. Figure 12a shows three consecutive cycles of RhB degradation using NiO-NiFe2O4-rGO activated PMS. It could be seen that the degradation efficiency of RhB using NiO-NiFe2O4-rGO was comparable with that using pristine NiO-NiFe2O4-rGO, even though NiO-NiFe2O4-rGO had not undergone any regeneration treatments. Even after three cycles, the degradation efficiency remained consistent without noticeable loss. Figure 12b shows the hysteresis loop of NiO-NiFe2O4-rGO, which was measured at room temperature with a maximum applied field from −20 to 20 kOe. The saturation magnetization of NiO-NiFe2O4-rGO was determined to be 25.17 emu/g, which was strong enough to separate from the solution via an external magnet (the inset of Figure 12a). Since the NiO-NiFe2O4-rGO catalyst was a highly magnetic material, the recovery of NiO-NiFe2O4-rGO catalyst from water could be easily implemented by placing an external magnet (neodymium) to keep the NiO-NiFe2O4-rGO catalyst within the beaker and then the aqueous solutions could be removed from the beaker. The weight changes before and after a recycle test was measured and it was found that the weight change (dry basis) was less than 1 wt%. Thus, no additional NiO-NiFe2O4-rGO catalyst was required for a subsequent cyclic test. These recyclability tests revealed that the NiO-NiFe2O4-rGO catalyst could be re-usable.
4. Conclusions
In this study, we demonstrated that NiO-NiFe2O4-rGO was an effective catalyst for the activation of peroxymono-sulfate (PMS) to efficiently degrade RhB in an aqueous solution. Elevated temperature and pH improved RhB degradation efficiency by using NiO-NiFe2O4-rGO activated PMS. The presence of HCO3− and Cl− were found to facilitate the removal of RhB. However, the addition of HPO42− noticeably hindered the degradation process. Through quenching experiments and EPR spectroscopic analyses, NiO-NiFe2O4-rGO was proven to activate PMS to generate SO4−· and OH·. The facile synthesis of the catalyst, along with its stability and the simple experimental procedure, hold promise for the investigation of the NiO-NiFe2O4-rGO/PMS system for the decomposition of persistent organic chemicals. We believe that these results may open up a new avenue for the design and preparation of various novel heterogeneous catalysts for PMS activation in the advanced oxidation processes.
Author Contributions
Author Contributions: Conceptualization, X.X. and F.L.; Methodology, X.X. and F.L.; Formal Analysis, X.X.; Investigation, F.L.; Resources, F.L.; Data Curation, F.L.; Writing-Original Draft Preparation, X.X.; Writing-Review and Editing, Q.Z.; Visualization, P.H.; Supervision, Q.Z. and F.Y.; Project Administration, F.Y.
Funding
This research was funded by the National Key Scientific and Technological Project for Water Pollution Control and Management (2012ZX07202-002).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bili´nskaa, L.; Gmurek, M.; Ledakowicz, S. Textile wastewater treatment by AOPs for brine reuse. Process Saf. Environ. 2017, 109, 420–428. [Google Scholar] [CrossRef]
- Rosario-Ortiz, F.L.; Wert, E.C.; Snyder, S.A. Evaluation of UV/H2O2 treatment for the oxidation of pharmaceuticals in wastewater. Water Res. 2010, 44, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.-A.; Hofmann, R. A comparative study of the hydroxyl radical scavenging capacity of activated sludge and membrane bioreactor wastewater effluents. Water Sci. Technol. 2016, 73, 2067–2073. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, F.; Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review. Chem. Eng. J. 2017, 310, 41–62. [Google Scholar] [CrossRef]
- Cheng, M.; Zeng, G.; Huang, D.; Lai, C.; Lui, Y.; Zhang, C.; Wan, J.; Hu, L.; Zhou, C.; Xiong, W. Efficient degradation of sulfamethazine in simulated and real wastewaterat slightly basic pH values using Co-SAM-SCS/H2O2 fenton-like system. Water Res. 2018, 138, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Mahdi-Ahmed, M.; Chiron, S. Ciprofloxacin oxidation by UV-C activated peroxymonosulfate in wastewater. J. Hazard. Mater. 2014, 265, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Babuponnusami, A.; Muthukumar, K. A review on Fenton and improvements to the Fenton process for wastewater treatment. J. Environ. Chem. Eng. 2014, 2, 557–572. [Google Scholar] [CrossRef]
- Zhang, B.T.; Zhang, Y.; Teng, Y.H.; Fan, M.H. Sulfate radical and its application in decontamination technologies. Crit. Rev. Env. Sci. Tec. 2015, 45, 1756–1800. [Google Scholar] [CrossRef]
- Fan, G.; Li, F.; Evans, D.G. Catalytic applications of layered double hydroxides: Recent advances and perspectives. Chem. Soc. Rev. 2014, 43, 7040–7066. [Google Scholar] [CrossRef] [PubMed]
- Monaam, B.; Alexandre, B.; Ahmed, A.; Brigitte, S.; Habib, E.; Mokhtar, F.; Sabine, S.; Rabah, B. Co2SnO4 nanoparticles as a high performance catalyst for oxidative degradation of rhodamine B dye and pentachlorophenol by activation of peroxymonosulfate. Phys. Chem. Chem. Phys. 2017, 19, 6569–6578. [Google Scholar]
- Ren, Y.; Lin, L.; Ma, J.; Yang, J.; Feng, J.; Fan, Z. Sulfate radicals induced from peroxymonosulfate by magnetic ferro spinel MFe2O4(M = Co, Cu Mn, and Zn) as heterogeneous catalysts in the water. Appl. Catal. B 2015, 165, 572–578. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Li, M.; Fan, J.; Zhao, G. Magnetic ordered mesoporous copper ferrite as a heterogeneous Fenton catalyst for the degradation of imidacloprid. Appl. Catal. B 2014, 147, 534–545. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, Y.; Lu, F.; Wei, F.; Wang, X.; Wang, S. Magnetic recoverable MnFe2O4 and MnFe2O4-graphene hybrid as heterogeneous catalysts of peroxymonosulfate activation for efficient degradation of aqueous organic pollutants. J. Hazard. Mater. 2014, 270, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.J.; Chu, W.; Gan, L. Environmental application of graphene-based CoFe2O4 as an activator of peroxymonosulfate for the degradation of a plasticizer. Chem. Eng. J. 2015, 263, 435–443. [Google Scholar] [CrossRef]
- Hummers, W.S., Jr.; Offeman, R.E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Zong, M.; Huang, Y.; Ding, X. One-step hydrothermal synthesis and microwave electromagnetic properties of RGO/NiFe2O4 composite. Ceram. Int. 2014, 40, 6821–6828. [Google Scholar] [CrossRef]
- Fu, M.; Jiao, Q.; Zhao, Y. Preparation of NiFe2O4 nanorod-graphene composites via an ionic liquid assisted one-step hydrothermal approach and their microwave absorbing properties. J. Mater. Chem. A 2013, 1, 5577–5586. [Google Scholar] [CrossRef]
- Liao, Y.; Fu, M.; Chen, L.; Wu, J.; Huang, B.; Ye, D. Catalytic oxidation of toluene over nanorod-structured Mn–Ce mixed oxides. Catal. Today 2013, 216, 220–228. [Google Scholar] [CrossRef]
- Ball, D.L.; Edwards, J.O. The kinetics and mechanism of the decomposition of Caro’s acid. J. Am. Chem. Soc. 1956, 78, 1125–1129. [Google Scholar] [CrossRef]
- Rastogi, A.; Al-Abed, S.R.; Dionysiou, D.D. Sulfate radical-based ferrous–peroxymonosulfate oxidative system for PCBs degradation in aqueous and sediment systems. Appl. Catal. B 2009, 85, 171–179. [Google Scholar] [CrossRef]
- Hussain, I.; Zhang, Y.Q.; Huang, S.B. Degradation of aniline with zero-valent iron as an activator of persulfate in aqueous solution. Rsc Adv. 2014, 4, 3502–3511. [Google Scholar] [CrossRef]
- Yang, S.; Wang, P.; Yang, X.; Shan, L.; Zhang, W.; Shao, X.; Niu, R. Degradation efficiencies of azo dye Acid Orange 7 by the interaction of heat, UV and anions with common oxidants: Persulfate, peroxymonosulfate and hydrogen peroxide. J. Hazard. Mater. 2010, 179, 552–558. [Google Scholar] [CrossRef]
- Qi, C.D.; Liu, X.T.; Zhao, W.; Lin, C.Y.; Ma, J.; Shi, W.X.; Sun, Q.; Xiao, H. Degradation and dechlorination of pentachlorophenol by microwave-activated persulfate. Environ. Sci. Pollut. R. 2015, 22, 4670–4679. [Google Scholar] [CrossRef]
- Qi, F.; Chu, W.; Xu, B. Modeling the heterogeneous peroxymonosulfate/Co-MCM41 process for the degradation of caffeine and the study of influence of cobalt sources. Chem. Eng. J. 2014, 235, 10–18. [Google Scholar] [CrossRef]
- Ji, Y.; Dong, C.; Kong, D.; Lu, J. New insights into atrazine degradation by cobalt catalyzed peroxymonosulfate oxidation: Kinetics, reaction products and transformation mechanisms. J. Hazard. Mater. 2015, 285, 491–500. [Google Scholar] [CrossRef]
- Chan, K.H.; Chu, W. Degradation of atrazine by cobalt-mediated activation of peroxymonosulfate: Different cobalt counteranions in homogenous process and cobalt oxide catalysts peroxymonosulfate. Water Res. 2009, 43, 2513–2521. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Ang, H.M.; Tade, M.O.; Wang, S. Facile Synthesis of hierarchically structured magnetic MnO2/ZnFe2O4 hybrid materials and their performance in heterogeneous activation of peroxymonosulfate. ACS Appl. Mater. Interfaces 2014, 6, 19914–19923. [Google Scholar] [CrossRef]
- Wu, T.X.; Liu, G.M.; Zhao, J.C. Photoassisted degradation of dye pollutants. V. Self-photosensitized oxidative transformation of Rhodamine B under visible light irradiation in aqueous TiO2 dispersions. J. Phys. Chem. 1998, 102, 5845–5851. [Google Scholar] [CrossRef]
Figure 1.
X-ray diffraction (XRD) pattern of NiO-NiFe2O4 and NiO-NiFe2O4-rGO.

Figure 2.
(a,b) Sanning electron microscopy (SEM) images and (c–f) elemental mapping of NiO-NiFe2O4-rGO.
Figure 2.
(a,b) Sanning electron microscopy (SEM) images and (c–f) elemental mapping of NiO-NiFe2O4-rGO.

Figure 3.
(a) Transmission electron microscope (TEM), image and (b) HRTEM image of NiO-NiFe2O4-rGO.

Figure 4.
(a) N2 adsorption-desorption isotherm. (b) Pore size distribution of NiO-NiFe2O4 and NiO-NiFe2O4-rGO.
Figure 4.
(a) N2 adsorption-desorption isotherm. (b) Pore size distribution of NiO-NiFe2O4 and NiO-NiFe2O4-rGO.

Figure 5.
(a) X-ray photoelectron spectroscopy (XPS )spectrum, (b) Ni 2p XPS spectrum, (c) Fe 2p XPS spectrum, (d) O 1s XPS spectrum of NiO-NiFe2O4, and NiO-NiFe2O4-rGO.
Figure 5.
(a) X-ray photoelectron spectroscopy (XPS )spectrum, (b) Ni 2p XPS spectrum, (c) Fe 2p XPS spectrum, (d) O 1s XPS spectrum of NiO-NiFe2O4, and NiO-NiFe2O4-rGO.

Figure 6.
The degradation of (a) rhodamine B (RhB) concentration and (b) total organic carbon (TOC) in different systems.
Figure 6.
The degradation of (a) rhodamine B (RhB) concentration and (b) total organic carbon (TOC) in different systems.

Figure 7.
Effects of (a) NiO-NiFe2O4-rGO dosage, (b) peroxymono-sulfate (PMS) dosage, (c) initial pH, and (d) temperature on the degradation of RhB.
Figure 7.
Effects of (a) NiO-NiFe2O4-rGO dosage, (b) peroxymono-sulfate (PMS) dosage, (c) initial pH, and (d) temperature on the degradation of RhB.

Figure 8.
The effects of different anions (a) NO3−, (b) H2PO4−, (c) HCO3−, and (d) Cl− on the degradation of RhB.
Figure 8.
The effects of different anions (a) NO3−, (b) H2PO4−, (c) HCO3−, and (d) Cl− on the degradation of RhB.

Figure 9.
(a) The effect of radical scavengers on the degradation of RhB; (b) EPR spectroscopic analyses.
Figure 9.
(a) The effect of radical scavengers on the degradation of RhB; (b) EPR spectroscopic analyses.

Figure 10.
HPLC chromatograms of RhB and its intermediate products in photocatalytic degradation for different reaction time: (a) 0 min; (b) 10 min; (c) 20 min; (d) 40 min.
Figure 10.
HPLC chromatograms of RhB and its intermediate products in photocatalytic degradation for different reaction time: (a) 0 min; (b) 10 min; (c) 20 min; (d) 40 min.

Figure 11.
The degradation pathways for RhB.

Figure 12.
(a) Recyclability of NiO-NiFe2O4-rGO to activate PMS; (b) The magnetic hysteresis loops of NiO-NiFe2O4-rGO magnetic nanomaterials.
Figure 12.
(a) Recyclability of NiO-NiFe2O4-rGO to activate PMS; (b) The magnetic hysteresis loops of NiO-NiFe2O4-rGO magnetic nanomaterials.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Structural properties of NiO-NiFe2O4 and NiO-NiFe2O4-rGO.
| Sample | SBET (m2·g−1) | Vtotal (cm3·g−1) | Vmic (cm3·g−1) |
|---|---|---|---|
| NiO-NiFe2O4 | 56.29 | 0.36 | 0.005 |
| NiO-NiFe2O4-rGO | 70.6 | 0.43 | 0.004 |
Table 2.
The effect of temperature on reaction rate.
| Temperature (K) | ||||
| 293 | 303 | 313 | 323 | |
| K (min−1) | 0.3012 | 0.2231 | 0.1423 | 0.1003 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xu, X.; Li, Y.; Zhang, G.; Yang, F.; He, P. NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B. Water 2019, 11, 384. https://doi.org/10.3390/w11020384
AMA Style
Xu X, Li Y, Zhang G, Yang F, He P. NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B. Water. 2019; 11(2):384. https://doi.org/10.3390/w11020384
Chicago/Turabian StyleXu, Xiaochen, Yanfang Li, Guoquan Zhang, Fenglin Yang, and Ping He. 2019. "NiO-NiFe2O4-rGO Magnetic Nanomaterials for Activated Peroxymonosulfate Degradation of Rhodamine B" Water 11, no. 2: 384. https://doi.org/10.3390/w11020384
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.