Article Text

Abstract
Background The role of club cells in the pathology of idiopathic pulmonary fibrosis (IPF) is not well understood. Protein disulfide isomerase A3 (PDIA3), an endoplasmic reticulum-based redox chaperone required for the functions of various fibrosis-related proteins; however, the mechanisms of action of PDIA3 in pulmonary fibrosis are not fully elucidated.
Objectives To examine the role of club cells and PDIA3 in the pathology of pulmonary fibrosis and the therapeutic potential of inhibition of PDIA3 in lung fibrosis.
Methods Role of PDIA3 and aberrant club cells in lung fibrosis was studied by analyses of human transcriptome dataset from Lung Genomics Research Consortium, other public resources, the specific deletion or inhibition of PDIA3 in club cells and blocking SPP1 downstream of PDIA3 in mice.
Results PDIA3 and club cell secretory protein (SCGB1A1) signatures are upregulated in IPF compared with control patients. PDIA3 or SCGB1A1 increases also correlate with a decrease in lung function in patients with IPF. The bleomycin (BLM) model of lung fibrosis showed increases in PDIA3 in SCGB1A1 cells in the lung parenchyma. Ablation of Pdia3, specifically in SCGB1A1 cells, decreases parenchymal SCGB1A1 cells along with fibrosis in mice. The administration of a PDI inhibitor LOC14 reversed the BLM-induced parenchymal SCGB1A1 cells and fibrosis in mice. Evaluation of PDIA3 partners revealed that SPP1 is a major interactor in fibrosis. Blocking SPP1 attenuated the development of lung fibrosis in mice.
Conclusions Our study reveals a new relationship with distally localised club cells, PDIA3 and SPP1 in lung fibrosis and inhibition of PDIA3 or SPP1 attenuates lung fibrosis.
- idiopathic pulmonary fibrosis
- interstitial fibrosis
Data availability statement
All the primary data are available from the authors upon request. Human data are available in the public databases that are documented in the manuscript.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is the key question?
What is the role of club cells and protein disulfide isomerase A3 (PDIA3) in pulmonary fibrosis pathology?
What is the bottom line?
Our study demonstrates that PDIA3 and profibrotic growth factor osteopontin are increased in idiopathic pulmonary fibrosis (IPF) and correlates with lung function decline in IPF. Similar increases in fibrotic mice were found, and inhibition or ablation of PDIA3 or osteopontin decreased the aberrant club cell population and lung fibrosis in mice.
Why read on?
This report identifies PDIA3 and osteopontin as potential drug targets in lung fibrosis, and blocking SPP1 or inhibiting PDIA3 attenuates lung fibrosis development in mice.
Introduction
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease that remains without effective therapeutics and elusive mechanisms of pathogenesis. The epithelium is the original site of lung injury, and repeated alveolar epithelial damage is often associated with progenitor cell dysfunction, abnormal repair and chronic fibrosis.1 2 Among these progenitor cells are non-ciliated club cells in the airway epithelium that perform several functions, including immunomodulation, maintaining homeostasis and regenerating bronchiolar walls.3 4 There is increasing evidence that club cells play essential roles in lung repair and found in the distal lung in IPF.5–8 However, the role of these distal club cells in lung fibrosis is not well studied.
Protein disulfide isomerases (PDIs) are an endoplasmic reticulum (ER)-based redox chaperones, which catalyse the formation or isomerisation of disulfide bonds (–S–S–) in proteins. PDIA3, a unique member of the PDI family of proteins, is primarily involved in the redox modification of newly synthesised glycoproteins and is upregulated during ER stress.9 PDIA3 has been implicated in diverse human diseases.10–12 However, the role of PDIA3 in pulmonary fibrosis is not fully elucidated. PDIA3 is known to regulate the profibrotic growth factor osteopontin (SPP1) production.13 Whether SPP1 is involved in the pathology of pulmonary fibrosis in conjunction with PDIA3 is not well known. Therefore, our objective was to examine the relationship of club cells, PDIA3 and SPP1 in the pathogenesis of pulmonary fibrosis and the therapeutic potential of the inhibition of PDIA3 in fibrosis.
In the present study, we observed a significant increase in PDIA3, including SCGB1A1 and SPP1 in IPF. Increases in expression of PDIA3, SCGB1A1 and SPP1 correlated with a significant decline in lung function in patients with IPF. Time-course analysis of bleomycin (BLM)-challenged mouse lung samples revealed club cell signature at the distal part of the lung, including increases in PDIA3 in the lung parenchyma and increased production of SPP1. Our results further show that club cell-specific Pdia3 ablation attenuated lung fibrosis in BLM-challenged mice. Similarly, the therapeutic administration of PDI inhibitor, LOC14, decreased fibrosis and club cell signature in the lung parenchyma. Next, by immunoprecipitation of PDIA3 and subsequent proteomics analysis, we identified profibrotic growth factor, SPP114 15 as an interactor of PDIA3 in fibrotic mice. Treatment with SPP1 antagonistic antibody in BLM-challenged mice decreased fibrosis. This study suggests that PDIA3 and SPP1 inhibition could be potential therapeutic targets in pulmonary fibrosis. Keeping in view the significance of club cells in lung repair and role of Pdia3 in pulmonary fibrosis, the objective of this study is to understand the function of club cells in the pathology of pulmonary fibrosis and mechanisms of action of PDIA3 in this disease.
Materials and methods
Detailed methods are described in the online supplemental materials and methods.
Supplemental material
Human samples
Deidentified frozen lung tissue from the patients diagnosed with IPF and control were obtained from the National Institutes of Health Lung Tissue Research Consortium (LTRC). Patient cohorts from the publicly available GSE47460 (microarray)16 and GSE150910 (bulk RNASeq)17 datasets were used to determine differential gene expression. Analyses were conducted on existing de-identified archived samples. Therefore the University of Vermont institutional review board no. M13-306, determined that the study is non-human subject research.
Animals
C57BL/6NJ male mice (Jackson Laboratories, Bar Harbor, ME, USA) aged around 12 weeks were studied. Mice were oropharyngeally instilled with 2.0 U/kg BLM (TEVA generic, Parsippany, NJ, USA), and the experimental control mice received phosphate-buffered saline (PBS). For the time-course studies, mice were harvested at day 4, 10, 14, 18 or 24, and bronchoalveolar lavage fluid for total and differential cell counts and protein analysis, the right upper lobe for hydroxyproline quantification, and the rest of the two lobes for western blot, immunoprecipitation, and RT-qPCR analysis, and the left lung for histologic analysis were collected. Mouse studies described herein were approved by IACUC protocol numbers PROTO 0201900016 and 0202000102.
Statistics
All mice studies were repeated once. All statistical analysis was carried out using Graph Pad Prism 8. The ROUT18 method was used to identify outliers with a cut-off of Q=2%, and identified outliers were removed from the subsequent statistical analysis (see figure legends). Data were pooled from two experiments and analysed by one or two-way analysis of variance where appropriate and a two-stage linear step-up procedure of Benjamini et al 19 (false discovery rate, Q=5% or 0.05) test to adjust for multiple comparisons. Adjusted p values (‘q’ values) of <0.05 were regarded as discovery in FDR. Mann-Whitney (for human data) and parametric Student’s t-test were used where appropriate. P values <0.05 were regarded as statistically significant in Mann-Whitney and parametric Student’s t-test. Data for all the results were expressed as ±SEM.
Results
Increased expression of PDIA3 in the lungs of patients with IPF
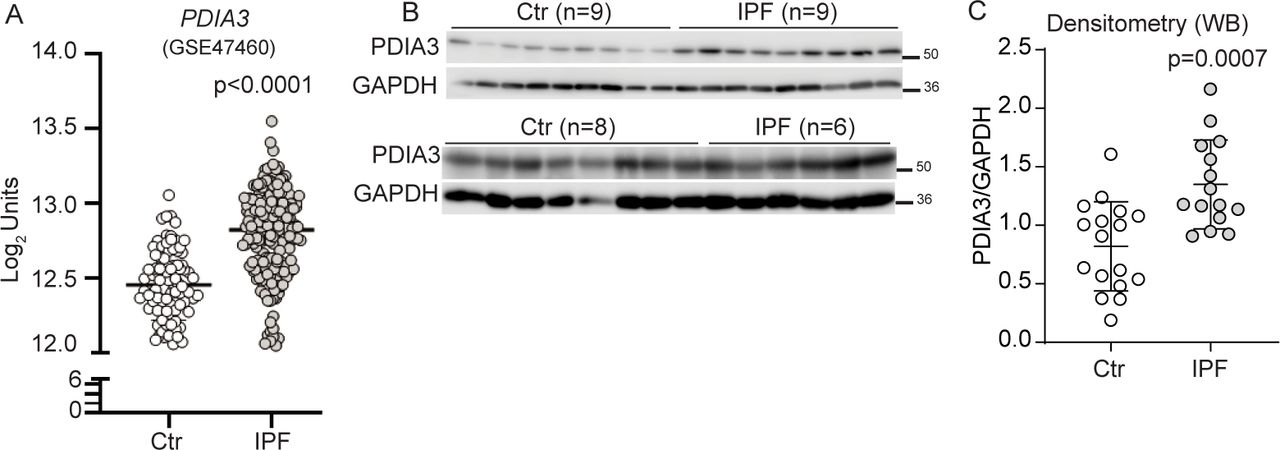
Retrospective analyses of the publicly available databases (GSE4746016 and GSE15091017) and LTRC samples showed that several PDIA mRNAs, including PDIA3 (protein), were significantly upregulated in patients with IPF as compared with controls (figure 1A–C, and online supplemental figure S1A, B). A significant correlation between PDIA3 increases in expression and a decline in the percent predicted diffusing capacity for lung carbon monoxide (% DLCO) and forced vital capacity (FVC) in patients with IPF was observed (online supplemental figure S1C–E). Along with increases in PDIA3 expression, significant increases in ER stress-dependent genes such as HSPA5, HSP90B1 and XBP1 in patients with IPF as compared with controls (online supplemental figure S1F–H) were also observed. These results indicate that the patients with IPF show elevated levels of ER stress markers, along with PDIA3. Patient demographic data of patients with non-IPF and IPF from LTRC and Lung Genomics Research Consortium are presented in online supplemental tables S4 and S5.
Supplemental material
Supplemental material
Supplemental material

Elevated expression of PDIA3 and SCGB1A1 in patients with IPF. (A) PDIA3 mRNA levels in control (n=132) and patients with IPF (n=160). (B) and (C) Western blot and quantitation of PDIA3 protein expression, control (n=17) and patients with IPF (n=15). Significance was based on unpaired two-tailed t-test and non-parametric Spearman correlation (rs) non-parametric Mann-Whitney t-test. Error bars represent ±SEM for the densitometry and ±SD for the mRNA analysis. GAPDH, glyceraldehyde phosphate dehydrogenase; IPF, idiopathic pulmonary fibrosis; PDIA3, protein disulfide isomerase A3.
PDIA3 and SCGB1A1 are upregulated in BLM-induced experimental lung fibrosis in mice
To study the implications of increases in PDIA3 in lung fibrosis, BLM was instilled via oropharyngeal aspiration into the mouse lungs, and the lungs were harvested at different time points (figure 2A). BLM-instilled mouse lungs showed a time-dependent increase in collagen deposition (on days 14, 18 and 24) compared with the lungs of PBS-instilled or BLM-instilled and harvested at day 4 (figure 2B). Quantitative RT-qPCR of unfolded protein response markers (Hspa5 and Xbp1) and Western blot analysis of PDIA3 revealed a significant upregulation of the genes mentioned above and proteins in a time-dependent manner in BLM-instilled mice compared with PBS-instilled mice (figure 2C,D and online supplemental figure S2A,B). The SCGB1A1 protein is specifically produced by club cells that are among the progenitor cell populations in the bronchioles. The SCGB1A1 gene signature is known to increase in pulmonary injury models and patients with IPF.4 20–22 Immunofluorescence and immunohistochemical staining of serial mouse lung tissue sections showed an increase and co-localisation of SCGB1A1 and PDIA3 in the parenchyma of fibrotic lungs. In contrast, the PBS-instilled lungs showed that SCGB1A1 and PDIA3 are primarily localised in the bronchiolar region (figure 2E,F and online supplemental figure S2C). Western blot analysis for SCGB1A1 in whole lung lysates also showed a significant time-dependent increase in BLM-instilled mouse lungs (figure 2G,H). Retrospective analyses of the publicly available gene expression datasets (GSE47460 and GSE150910)16 17 from lungs of patients with IPF showed that SCGB1A1 including isoforms SCGB3A1 and SCGB3A2 mRNAs were significantly increased in patients with IPF compared with controls (figure 2I and online supplemental figure S2D–F). A significant correlation between increases in SCGB1A1/SCGB3A1 expression and a decline in % DLCO and FVC (SCGB3A1) in patients with IPF was observed (online supplemental figure S2G–L). These results indicated that while SCGB1A1 and PDIA3 are normally expressed in the bronchiolar epithelial cells, following BLM-induced lung fibrosis, the SCGB1A1 and PDIA3 proteins, including co-localisations are increased in the lung parenchyma of fibrotic mice. Furthermore, lung function parameters negatively correlated with increases in expression of SCGB1A1 or other isoforms in patients with IPF.
Supplemental material

PDIA3 and SCGB1A1 are increased in the parenchyma in a mouse model of fibrosis. (A) Bleomycin (BLM) or PBS challenge and harvest regimen. (B) Time-dependent alterations in hydroxyproline content *p<0.05 as compared with 24-day PBS and #p<0.05 as compared with 4-day BLM samples by ANOVA; error bars±SEM (n=5–9 mice/group). (C, D) Western blot analysis and quantitation of PDIA3 normalised to the expression of β-actin in the lung lysates; *p<0.05 compared with 24-day PBS and #p<0.05 as compared with 4-day BLM samples by ANOVA (n=3 mice/group); error bars±SEM. (E and F) Representative images from confocal microscopy stained for SCGB1A1 (green), PDIA3 (red) and nucleus (blue) and quantitation of SCGB1A1 and PDIA3 in the parenchyma. Secondary antibody (without primary) staining on fibrotic mouse lungs is used as the negative control. Unpaired t-test, (n=3 mice/group); error bars±SEM. Scale bar 50 µm. (G, H) Western blot analysis and quantitation of SCGB1A1 normalised to the expression of β-actin in the lung lysates; *p<0.05 compared with 24-day PBS and #p<0.05 as compared with 4-day BLM samples by ANOVA (n=3 mice/group); error bars±SEM. (I) SCGB1A1 mRNA levels in control (n=132) and patients with IPF (n=160). ANOVA, analysis of variance; PBS, phosphate-buffered saline; PDIA3, protein disulfide isomerase A3.
Ablation of Pdia3 in SCGB1A1+ epithelial cells attenuates BLM-induced SCGB1A1 in lung parenchyma and fibrosis in mice
We reported earlier that the PDIA3 is required for allergen-induced airways fibrotic responses and apoptosis regulation by Fas in lung fibrosis.23 24 Here we observed that PDIA3 is upregulated in fibrotic mice and localises with SCGB1A1+ cells, including increases in mRNAs of SCGB1A1 (and other isoforms) in patients with IPF (figure 2). Next, we sought to determine whether an increase in PDIA3 in SCGB1A1 cells contribute to the pathology of lung fibrosis. We used a doxycycline-inducible transgenic Scgb1a1-rtTA/TetOP-Cre/Pdia3loxp/loxp mice (designated as ΔEpi-Pdia3).23 Littermate mice carrying Scgb1a1rtTA/TetOP-Cre or Scgb1a1-rtTA/Pdia3loxp/loxp were used as controls (designated as Ctr). Doxycycline-induced ablation of Pdia3 in lung epithelial cells of these mice has been characterised in our previous studies.23 25 To ensure the ablation of Pdia3 during the fibrotic phase (figure 2B), all mice were provided with a doxycycline diet 14 days after BLM or PBS installation for the rest of the experimental period (figure 3A). We found that BLM-induced fibrosis was significantly attenuated in ΔEpi-Pdia3 mice, reflected by decreased lung hydroxyproline levels (figure 3B). Analyses of fibrotic markers (Col1A1 and Fn-1) by RT-qPCR using selected primers (online supplemental table S7) and collagen deposition by Masson’s trichrome (MT) staining also indicated a significant decrease in BLM-induced fibrosis in ΔEpi-Pdia3 mice when compared with Ctr mice (figure 3C–F). Immunofluorescence staining of lung tissue sections of BLM-challenged ΔEpi-Pdia3 mice showed a significant decrease in SCGB1A1 staining in the parenchyma as compared with fibrotic Ctr mice (figure 3G,H). Ctr or ΔEpi-Pdia3 mice challenged with PBS did not show any alterations in the measurements reported above. Doxycycline-fed PBS-challenged mice (ΔEpi-Pdia3 mice) showed a significant decrease in PDIA3 staining in the mouse lung as compared with PBS challenged Ctr mice (online supplemental figure S3A,B). These results suggest that Pdia3 in club cells play an essential role in BLM-induced presence of SCGB1A1+ cells in the lung parenchyma, and the ablation of Pdia3 attenuates lung fibrosis potentially by diminishing SCGB1A1+ cells in the lung parenchyma.
Supplemental material
Supplemental material

Pdia3 ablation in SCGB1A1 cells decreases lung fibrosis in mice. (A) BLM or PBS-instillation, doxycycline treatment and harvest regimen. (B) Collagen content in the upper right lung lobe is measured by hydroxyproline content (n=5–11 mice/group). (C and D) Measurement of collagen and fibronectin mRNAs (n=5–11 mice/group). (E and F) Representative histochemical images of Masson’s trichrome (blue) stained lung sections and histological scoring (n=4–6 mice/group). (G and H) Representative confocal immunofluorescence images stained for SCGB1A1 and DAPI and quantitation of parenchymal SCGB1A1 (arrows) immunofluorescence (n=4 mice/group). *p<0.05 as compared with PBS and #p<0.05 as compared with Ctr-BLM samples by ANOVA; error bars±SEM. Arrows indicate SCGB1A1 immunoreactivity in the parenchyma. Scale bar 50 µm. Pooled samples from two experiments. ANOVA, analysis of variance; BLM, bleomycin; PBS, phosphate-buffered saline; PDIA3, protein disulfide isomerase A3.
Therapeutic administration of PDI inhibitor LOC14 decreases SCGB1A1+ cells and alleviates BLM-induced lung fibrosis
Our study showed that ablation of Pdia3 in club cells protects mice from BLM-induced fibrosis, including decrease in SCGB1A1+ cells (figure 3). Next, we explored whether PDIA3 inhibition using an inhibitor, LOC14,26 could potentially attenuate fibrosis in mice. Our previous work has shown that LOC14 inhibits recombinant (r)PDIA3 at an IC50 of ~5 µM.25 Starting at day 14 post BLM instillation, a time we observed increases in collagen (figure 2), we administered various concentrations of LOC14/vehicle (dimethyl sulfoxide (DMSO)) (figure 4A). LOC14 at a concentration of 0.15 mg/kg of mouse weight effectively alleviated fibrosis, as shown by a significant decrease in collagen deposition (figure 4B). BLM-instilled mice treated with vehicle control and 0.015 mg/kg or 1.5 mg/kg of LOC14 did not attenuate collagen deposition in the mouse lungs (figure 4B). The mRNA analysis for Col1a1, Fn-1, and histological quantitation by MT staining revealed attenuation of lung fibrosis in mice treated with 0.15 mg/kg LOC14 (figure 4C–F). BLM-challenged DMSO-treated mice showed a significant increase in Scgb1a1 and various Pdia mRNAs, including ER stress markers Hspa5 and Xbp1 that were attenuated in BLM-challenged, LOC14-treated lungs (figure 4G and online supplemental figure 3C–I). SCGB1A1 staining in the parenchyma compared with fibrotic mice was also significantly decreased in BLM-challenged, LOC14-treated mouse lungs (figure 4H,I). Furthermore, inflammatory cells (macrophages) were significantly decreased in BLM-instilled LOC14-treated (0.015/0.15 mg/kg) mice (online supplemental figure 4A–E). Mice instilled with PBS did not show any alterations in the measurements reported above. Measurement of liver injury marker ALT was not significantly altered in the groups reported above (online supplemental figure 4F). These results suggest that a PDI inhibitor LOC14 decreases the population of SCGB1A1+ cells in the lung parenchyma and also attenuates BLM-induced lung fibrosis.
Supplemental material

LOC14 decreases SCGB1A1 cells and lung fibrosis in mice. (A) BLM or PBS challenge, LOC14 treatment and harvest regimen (0.015, 0.15 and 1.5 mg/kg LOC14 were administered into the lung via oropharyngeal aspiration). (B) Collagen content in the upper right lung lobe, measured by hydroxyproline content (n=5–17 mice/group). (C and D) Measurement of collagen and fibronectin mRNAs (n=5–7 mice/group). (E and F) Representative histochemical images of Masson’s trichrome (blue) stained lung sections and histological scoring (n=5 mice/group). (G) Measurement of Scgb1a1 mRNA (n=5–7 mice/group). (H and I) Representative confocal immunofluorescence images stained for SCGB1A1 and DAPI and quantitation of parenchymal SCGB1A1 (arrows) immunofluorescence (n=4 mice/group). *p<0.05 as compared with PBS and #p<0.05 as compared with other-BLM samples by ANOVA; error bars±SEM. Arrows indicate SCGB1A1 immunoreactivity in the parenchyma. Scale bar 50 µm. Pooled samples from two experiments. ANOVA, analysis of variance; BLM, bleomycin; PBS, phosphate-buffered saline.
Identification of osteopontin (SPP1) as an interactor of PDIA3 in BLM-induced lung fibrosis
Aberrant lung epithelial cells produce growth factors that potentially enhance the course of fibrosis.27 Although PDIA3 is known to interact with various mediators of cellular processes, there is very little information regarding PDIA3 interacting proteins in the lung that contribute to fibrosis.23 28 29 Using immunoprecipitation and mass spectrometry-based proteomics approach, a total of 745 proteins, belonging to 548 families of proteins (clusters) were identified in PBS, BLM 14 day and BLM 24 day groups, with 70 protein clusters identified as differential interactors among the three groups (online supplemental tables S1–S3). Among the 548 protein clusters, 64 (a keratin cluster, the common contaminant is not counted) and 52 were differentially identified in BLM 14d (vs PBS) and BLM 24d (vs PBS), respectively (figure 5A,B and online supplemental figure 5A,B). The differentially interacting proteins (in triplicates, online supplemental figure S5C) were abundant in BLM-instilled PDIA3-immunoprecipitated groups compared with PBS controls, suggesting that these proteins specifically associate with PDIA3 in fibrotic mice. We identified a comprehensive set of PDIA3-interacting proteins, including profibrotic growth factors osteopontin (SPP1) and Galectin-315 ,14 (figure 5A,B). The PDIA3–SPP1 interaction was confirmed by immunoprecipitation of lung tissue lysates with anti-PDIA3 antibody and subsequent Western blotting. Time-dependent increases in SPP1 interactions with PDIA3 were observed on days 14 and 24 fibrotic mouse lung compared with PBS controls (figure 5C). Measurement of SPP1 in the bronchoalveolar lavage fluid (BALF) of fibrotic mice and lung tissue lysates of patients with IPF showed significant increases compared with PBS or controls (human), respectively (figure 5D,E). Retrospective analyses of the publicly available gene expression datasets (GSE47460 and GSE150910)16 17 also revealed that patients with IPF have a significant increase of SPP1 mRNA, which correlated with lung function decline (% DLCO and FVC) compared with controls (figure 5F,G and online supplemental figure S5D–F). These results indicate that PDIA3 interacts with various profibrotic proteins, including SPP1. Furthermore, SPP1 increases in a mouse model of fibrosis and IPF, along with lung function decline, suggests SPP1 as a potential modulator of lung fibrosis.
Supplemental material
Supplemental material
Supplemental material

Analysis of PDIA3 interacting partners and identification of SPP1 as a potential mediator of lung fibrosis. (A) Heat map of interacting partners of PDIA3 in fibrotic mouse lung analysed 14-day post-BLM challenge (n=3 samples/group, ‘C’ immunoprecipitation using non-specific IgG as a control). Identified proteins with a p value <0.05 (two-tailed t-test: 14-day post-BLM challenge vs PBS) are represented. The number beside the gene symbol indicates how many members in the cluster associated with that protein. The scale of the colour intensity is arbitrary. (B) Volcano plot depicting the significance of interactions of PDIA3 and SPP1 post immunoprecipitation and mass spectrometry. Fold changes of 2 and p value at 0.05 (two-tailed t-test: 24-day post-BLM challenge vs PBS) are indicated by dotted lines on the X-axis and y-axis, respectively. (C) Western blot analysis of immunoprecipitated PDIA3 and SPP1 in lung lysates (n=4 samples/group). (D) ELISA for SPP1 in bronchoalveolar lavage fluid samples of 24-day post-PBS (n=10) or BLM (n=12) challenged mice. (E) ELISA for SPP1 in lung tissue samples of control (n=18) and patients with IPF (n=16). (F) mRNA expression of SPP1 in control (n=132) and patients with IPF (n=160) from LGRC cohort. (G) Correlation of SPP1 mRNA expression with %DLCO in control (n=92) and patients with IPF (n=145) from LGRC cohort. Significance was based on unpaired two-tailed t-test and non-parametric Pearson correlation, non-parametric Mann-Whitney t-test. Error bars represent ±SEM for mouse samples and ±SD for human samples. ANOVA, analysis of variance; BLM, bleomycin; PBS, phosphate-buffered saline; PDIA3, protein disulfide isomerase A3.
Ablation or Inhibition of PDIA3 decreases SPP1 production, and blocking SPP1 attenuates lung fibrosis in mice
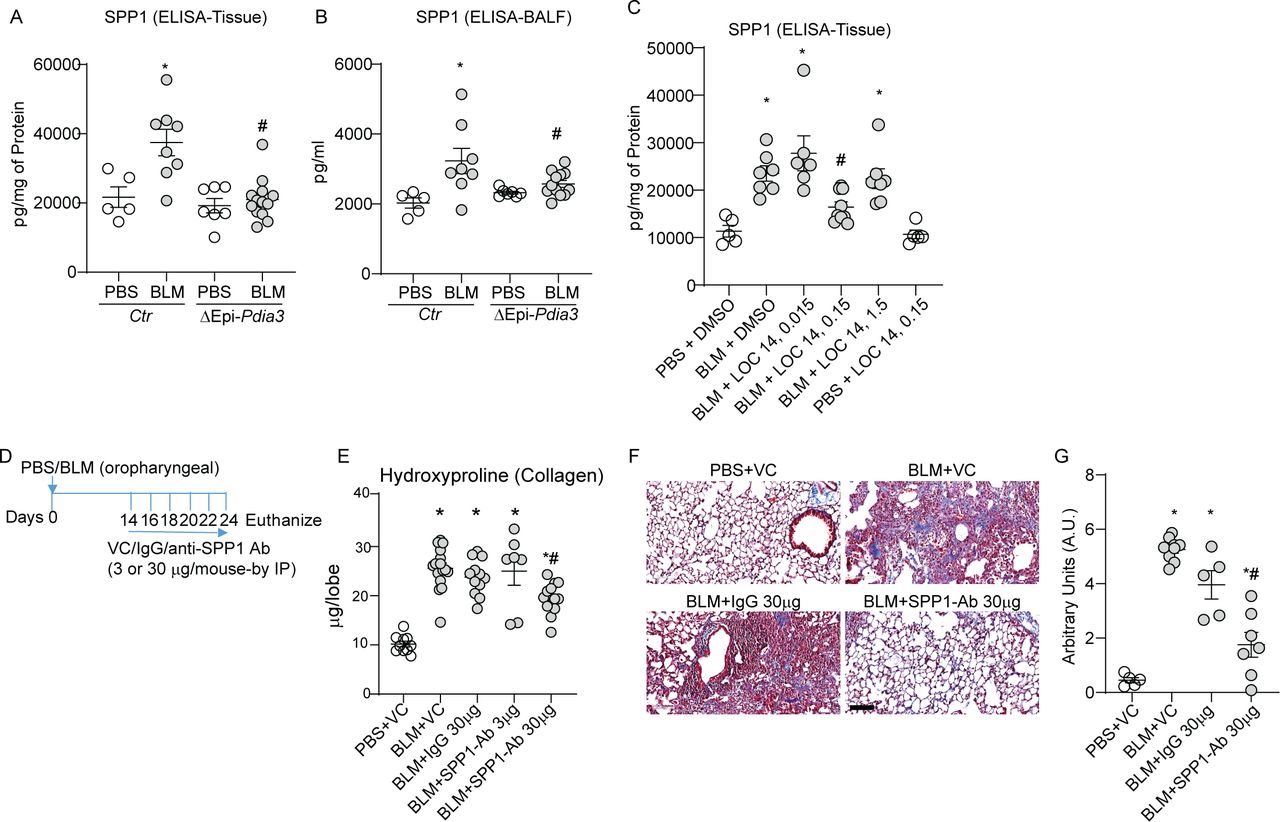
To further investigate the role of PDIA3 in SPP1 production and lung fibrosis, we analysed the lung tissue and BALF samples from our experiments in Ctr or ΔEpi-Pdia3 or LOC14 treated mice (figures 3 and 4). Measurement of SPP1 protein from the above experiments (in figures 3 and 4) showed a significant decrease in SPP1 levels in BLM challenged ΔEpi-Pdia3 or LOC14 (0.15 mg/kg) groups as compared with other BLM-challenged groups (figure 6A–C). These results suggest that the club cell-specific deletion or inhibition of PDIA3 decreases SPP1 production along with attenuation of lung fibrosis in mice.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Club cell-specific Pdia3 ablation or inhibition decreases SPP1 and SPP1 blocking antibody attenuates lung fibrosis in mice. (A, B) Measurement of SPP1 protein in lung tissue and BALF of Ctr and ΔEpi-Pdia3 mice. *p<0.05 as compared with PBS groups, #p<0.05 as compared with the Ctr-BLM group by ANOVA, error bars±SEM (n=5–13 mice/group). Outlier removed in BALFΔEpi-Pdia3 (n=1). (C) Measurement of SPP1 by ELISA in PBS or BLM challenged in combination with dimethyl sulfoxide or LOC14 treated mice. *p<0.05 as compared with PBS groups, #p<0.05 as compared with other-BLM groups by ANOVA, error bars±SEM (n=5–9 mice/group). (D) BLM or PBS challenge, VC/IgG/SPP1-Ab treatment and harvest regimen. (E) Collagen content in the upper right lobe is measured by hydroxyproline content (n=10–16 mice/group). (F and G) Representative histochemical images of Masson’s trichrome (blue) stained lung sections and histological scoring (n=5–8 mice/group). *p<0.05 as compared with PBS group and #p<0.05 as compared with other-BLM groups by ANOVA; error bars±SEM. Scale bar 400 µm. ANOVA, analysis of variance; BALF, bronchoalveolar lavage fluid; BLM, bleomycin; PBS, phosphate-buffered saline; PDIA3, protein disulfide isomerase A3.
To examine whether SPP1, downstream of PDIA3, is a target for lung fibrosis treatment, we blocked SPP1 using an antagonistic antibody (online supplemental table S6) 14 days after the BLM challenge (figure 6D). Lung collagen levels were significantly higher in BLM-instilled mice treated with the vehicle control, isotype IgG, or 3 µg of the anti-SPP1 antibody compared with PBS-vehicle control mice (figure 6E). The treatment of BLM-instilled mice with 30 µg of anti-SPP1 antibody significantly decreased collagen levels compared with all the other BLM groups (figure 6E). We also observed similar trends in MT staining of the lung tissue (figure 6F,G). PBS challenged, or vehicle-treated mice did not show any alterations in the reported measurements. Interestingly, SPP1 antibody-treated BLM-challenged mice did not show any decreases in inflammatory cell counts in the BALF (online supplemental figure 6A,B). Together, these results indicate that the SPP1, along with distally localised SCGB1A1+ cells downstream of PDIA3, promotes lung fibrosis in mice.
Supplemental material
Supplemental material
Discussion
ER stress and related proteins impact numerous cellular processes, including some that have been implicated in the pathogenicity of IPF.30 31 Our results demonstrate that PDIA3 and club cell signatures are increased in IPF lungs and these upregulations correlate with a significant decrease in lung function in patients with IPF. Targeted ablation or inhibition of PDIA3 in club cells decreases parenchymal club cells in the fibrotic mice. These studies also led to the discovery of profibrotic growth factor SPP1 as an interactor and downstream effector of PDIA3 in lung fibrosis. Furthermore, antagonistic SPP1 antibody treatment showed that blocking SPP1 attenuates lung fibrosis in mice.
Previous studies reported that aberrant club cells promote lung fibrosis and naphthalene-induced depletion of club cells attenuated lung fibrosis in mice.21 Injury to the bronchioles also induces the production of profibrotic mediators in the club cells.32 Abnormal distribution of club cells has been reported in IPF lungs.7 8 22 Interestingly, recent single-cell RNA seq and RNA in situ hybridisation studies also indicated the increases in club cell signature in the parenchyma of the patients with IPF.8 33 A recent study also suggested that progenitor SCGB1A1 niches are present in the alveolar regions and act as reservoirs for alveolar type II (ATII) epithelial cells during the injury and repair process of the lung.34 These reports suggest that expansion and overt responses by club cells potentially contribute to the pathology of the IPF. However, the functions or secretory profiles that induce this aberrant club cell contribution to profibrotic environment are not entirely clear. Although our study does not explore the origin and mode of expansion of the club cells in lung fibrosis, this work demonstrates that the aberrant club cells in the lung parenchyma associate with the production of profibrotic growth factor SPP1 and contributes to the perpetuation of fibrosis. Further, in-depth studies using single-cell multi-omics approaches, including organoid cultures, will provide greater insight into the active participation of club cells and their secretome in the pathogenicity of IPF.
PDIA3 facilitates oxidative folding (modifications) of various proteins, including the proteins that are mediators of pulmonary fibrosis (eg, Fas, Integrins and LOXL2).28 29 Systemic ablation of PDIA3 is embryonic lethal in mice, and the embryos do not progress beyond embryonic day 15, which coincides with lung, gut and brain development,35 and potentially suggesting a role for PDIA3 in lung development. We developed a mouse strain that conditionally deletes PDIA3 in SCGB1A1 positive (+) cells.23 Our analyses demonstrate that most of the PDIA3 expression was restricted to SCGB1A1+ cells in non-fibrotic mice’s bronchioles. However, in the fibrotic mice, PDIA3 co-localised with both bronchiolar and parenchymal SCGB1A1+ cells, indicating a pathological role for PDIA3 in fibrosis. Our earlier studies suggested a role for PDIA3 in allergen-induced peribronchial fibrosis and lung fibrosis.23 24 This report expands that knowledge to the aberrant SCGB1A1+ cells found in the parenchyma of fibrotic lungs. A previous study identified that PDIA3 is involved in ATII pneumocyte-to-AT I cell trans-differentiation, indicating a role for PDIA3 in alveolar epithelial cell differentiation.36 Our study did not explore the cell trans-differentiation, proliferation or migration, we found that the deletion of Pdia3 attenuated BLM-induced lung fibrosis, including decreases in population SCGB1A1+ cells in the lung parenchyma of fibrotic mice.
Our follow-up study with the therapeutic administration of a PDIA3 inhibitor, LOC14,25 also demonstrated a significant decrease in markers of fibrosis and SCGB1A1+ cells in fibrotic mice. These results further indicated that the PDIA3 potentially supports the SCGB1A1+ cells in the parenchyma and that inhibition or ablation of PDIA3 attenuates fibrosis in mice by diminishing the SCGB1A1+ cell population. Our results indicate that PDIA3 cell-specific deletion or targeted inhibition is potentially beneficial in the treatment of fibrosis. Intriguingly, a retrospective analysis of recent single-cell RNA-seq datasets (GSE13589333 and GSE13683137) revealed that although not statistically significant, an increasing trend of PDIA3 signature in association with ATII-epithelial and club cell signatures in patients with IPF (online supplemental figure 7A,B). Immunofluorescence stainings for proSP-C and PDIA3 on mouse lung tissue sections displayed co-localisation of proSP-C and PDIA3 in the parenchyma of PBS as well as in fibrotic mouse lungs (online supplemental figure 8A,B). The proSP-C signal showed an increasing trend but were not statistically significant in fibrotic mouse lungs compared with PBS. Lung tissues of patients with IPF and normal donors showed co-localisation signals of proSP-C and PDIA3 in the parenchyma of both IPF and normal chohorts, whereas of SCGB1A1 with PDIA3 was found to be co-localised mainly in the bronchioles of normal lung and in both the bronchioles and parenchyma in IPF lung (online supplemental figure 8C–F). However, none of these trends were statistically significant. These results suggest that PDIA3 potentially associates with both pro-SPC and SCGB1A1, however, further detailed studies in mice and in large set of patient samples are required to clarify the role of PDIA3 in these important epithelial population.
Supplemental material
Supplemental material
Aberrant activation of the epithelial cell population potentiates a fibrotic milieu in lung fibrosis.2 PDIA3 being a redox chaperone, could support folding and secretion of growth factors.23 Since the inhibition of PDIA3 decreases parenchymal SCGB1A1+ cells and fibrosis, we next explored PDIA3 interacting proteins in the fibrotic and control mouse lungs. Our analysis revealed, among other proteins, a significant increase in the interaction of PDIA3 with profibrotic growth factor SPP1 which also correlated with lung function decrease in patients with IPF.
Our results showed that club cell-specific ablation of PDIA3 decreased production of SPP1 in experimental fibrosis, indicating a direct role of PDIA3 in SPP1 production. SPP1 is a multifunctional protein expressed in activated macrophages, T-cells, osteoclasts, hepatocytes, smooth muscle, endothelial and epithelial cells.38 Interestingly, knockdown of PDIA3 also showed a decrease in the production of SPP1 in osteoblast-like MC3T3-E1 cells.13 The ablation of Spp1 has been shown to suppress nanoparticle-induced injury and lung fibrosis in mice.39
A recent study has identified a subpopulation of SPP1 high macrophages and club cells in the lower lobes of lungs of patient with IPF. Furthermore, suggesting that these macrophages are the major sources of SPP1 in IPF.40 Our studies reveal that an increase in club cells is associated with SPP1 production and pulmonary fibrosis. Although our study did not analyse the SPP1 in macrophages, based on the literature, we predict that future analysis will reveal a pathogenic regulatory loop among various cell types associated with SPP1 in IPF.
As introduced, PDIA3 functions as a disulfide altering enzyme, specifically in secreted glycoproteins.9 Our work does not confirm whether disulfides of SPP1 are altered in PDIA3 deleted or inhibited mice; further redox assays and cell fractionation studies, including cysteine labelling, are required to confirm alterations of SPP1 cysteine redox status. Recent reports also suggested SPP1 as a potential marker in various respiratory and cardiovascular diseases associated with fibrosis.14 41 Similar to PDIA3, a retrospective analysis of datasets (GSE13589333 and GSE13683137) revealed that although not statistically significant, an increasing trend in SPP1 among other cell types associating with club cell signatures in patients with IPF (online supplemental figure 7C,D). Interestingly, naphthalene-mediated depletion of club cells resulted in decrease in production of SPP1.31 Detailed studies are required to elucidate SPP1 receptors CD44, and α4β1 integrins42 that are abundantly expressed on various mesenchymal cells43 and their role in the SPP1 mediated profibrotic response in IPF.
Collectively, the present study indicates an increase in PDIA3 and SCGB1A1+ cells in the parenchyma, and the interaction of growth factor SPP1 with PDIA3 forms a pathological pro-fibrotic milieu. Importantly, we demonstrate that therapeutic phase inhibition of PDIA3 or SPP1 significantly attenuates lung fibrosis in mice.
Supplemental material
Data availability statement
All the primary data are available from the authors upon request. Human data are available in the public databases that are documented in the manuscript.
Ethics statements
Patient consent for publication
Acknowledgments
The authors thank excellent technical support from Ms Nicole Bufford and Ms Nicole DeLance of the University of Vermont Microscopy centre, and further financial support from the Department of Pathology and Laboratory Medicine, the University of Vermont.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AK and EE contributed equally.
Contributors AK, EE, YMWJ-H and VA conceived the study, performed the experiments, analysed, interpreted the data and wrote the manuscript. AK and EE conducted mouse experiments, analysed the samples and performed various biochemical, microscopy and other quantitative assays. SRB, ZFM, NC, JW, MR and BKM provided help in mouse harvest, processing of the lung tissues, BALF inflammatory cell analyses and ELISA. ZFM and BKM maintained the experimental mice and performed instillation/injection of various reagents. YWL, CG and RC provided support in performing mass spectrometry and analyses of the results. EE, JLG, AJ and SG analysed human microarray/RNA Seq data and correlated with patient characteristics.
Funding This work is supported by NIH R01s HL122383, HL141364 and HL 136917 to VA; R35 HL135828 to YMWJ-H; R01 HL153604 and R03 HL154275 to JLG; 1UG3TR002612 to AJ; T32 HL076122 fellowship to EE, SRB and NC and F31 HL144051 fellowship to EE. YWL and CG are supported by (INBRE programme) NIH P20GM103449.
Competing interests YMWJ-H and VA hold patents: United States Patent No. 8,679,811, 'Treatments Involving Glutaredoxins and Similar Agents', United States Patent No. 8,877,447, 'Detection of Glutathionylated Proteins' (to YMWJ-H), United States Patents, 9,907,828 and 10,688,150 'Treatments of oxidative stress conditions' (YMWJ-H and VA). In the past YMWJ-H and VA have received consulting fees and laboratory contracts from Celdara Medical LLC, NH.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Linked Articles
- Airwaves