
DFMO Improves Survival and Increases Immune Cell Infiltration in Association with MYC Downregulation in the Pancreatic Tumor Microenvironment

 and
and 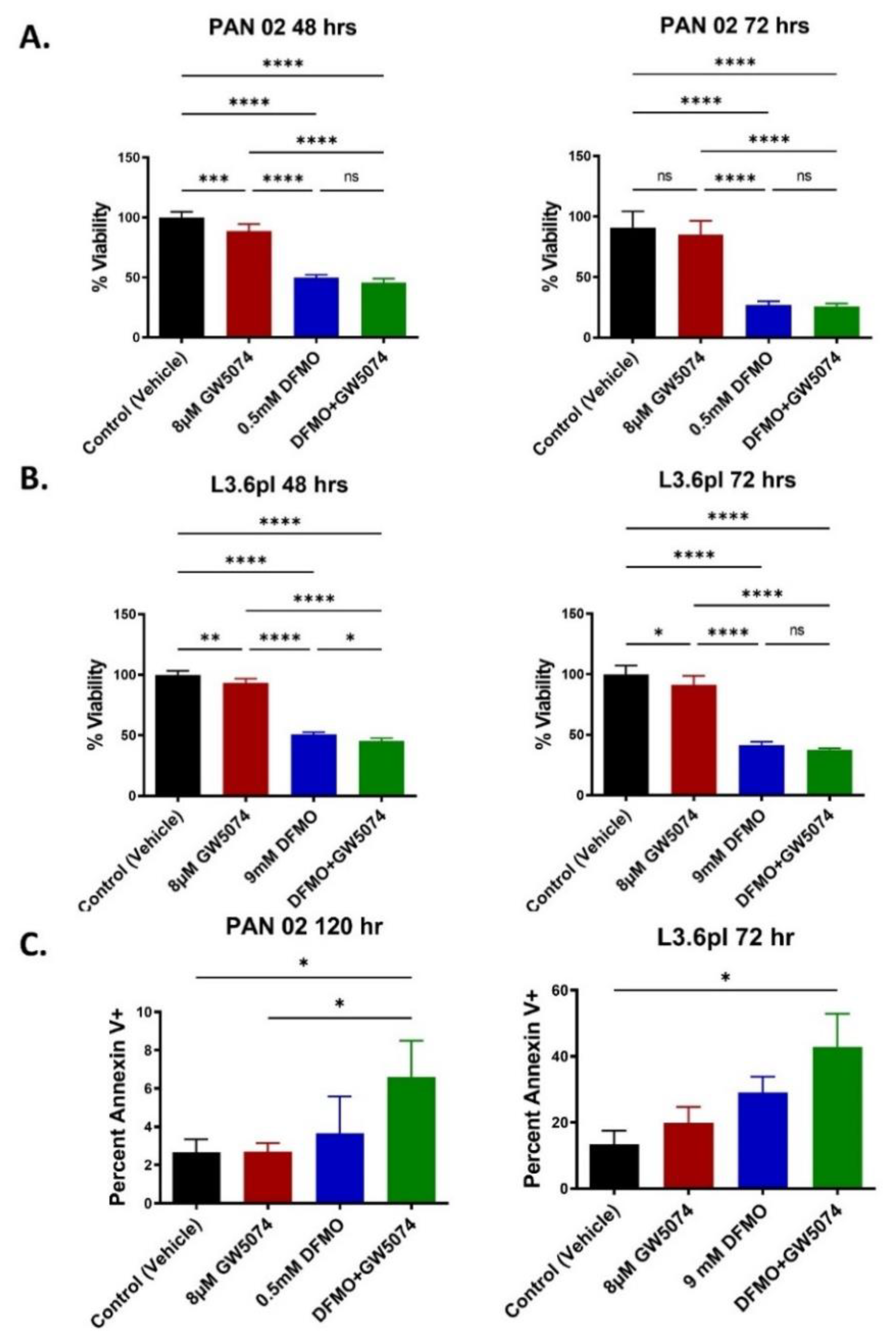{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. DFMO and DFMO + GW5074 Treatments Decrease Pancreatic Cancer Cell Viability In Vitro
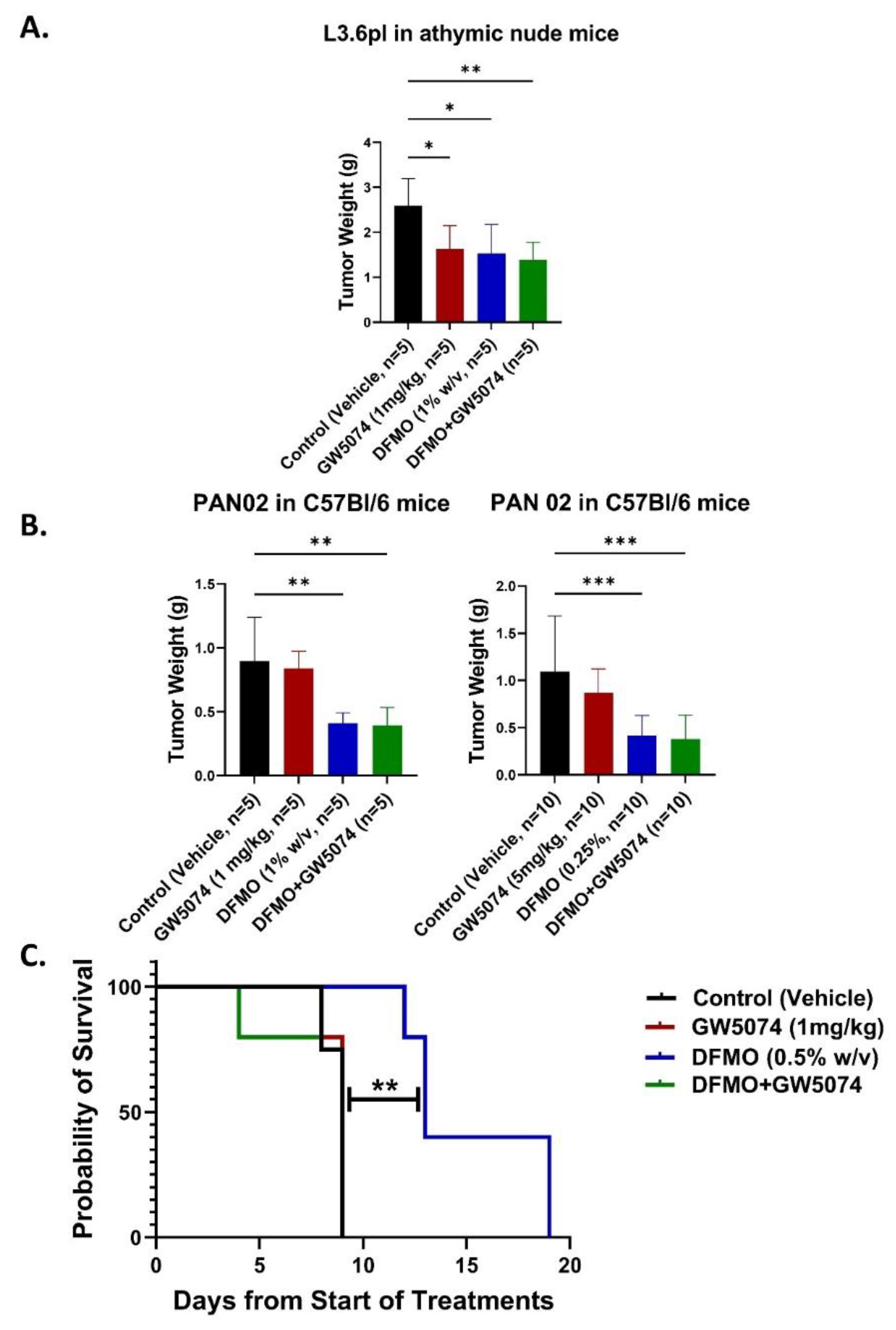
2.2. DFMO Alone Markedly Increases Survival of Pancreatic Tumor-Bearing Mice in Contrast with GW5074 across Multiple Dosing and Tumor Seeding Strategies
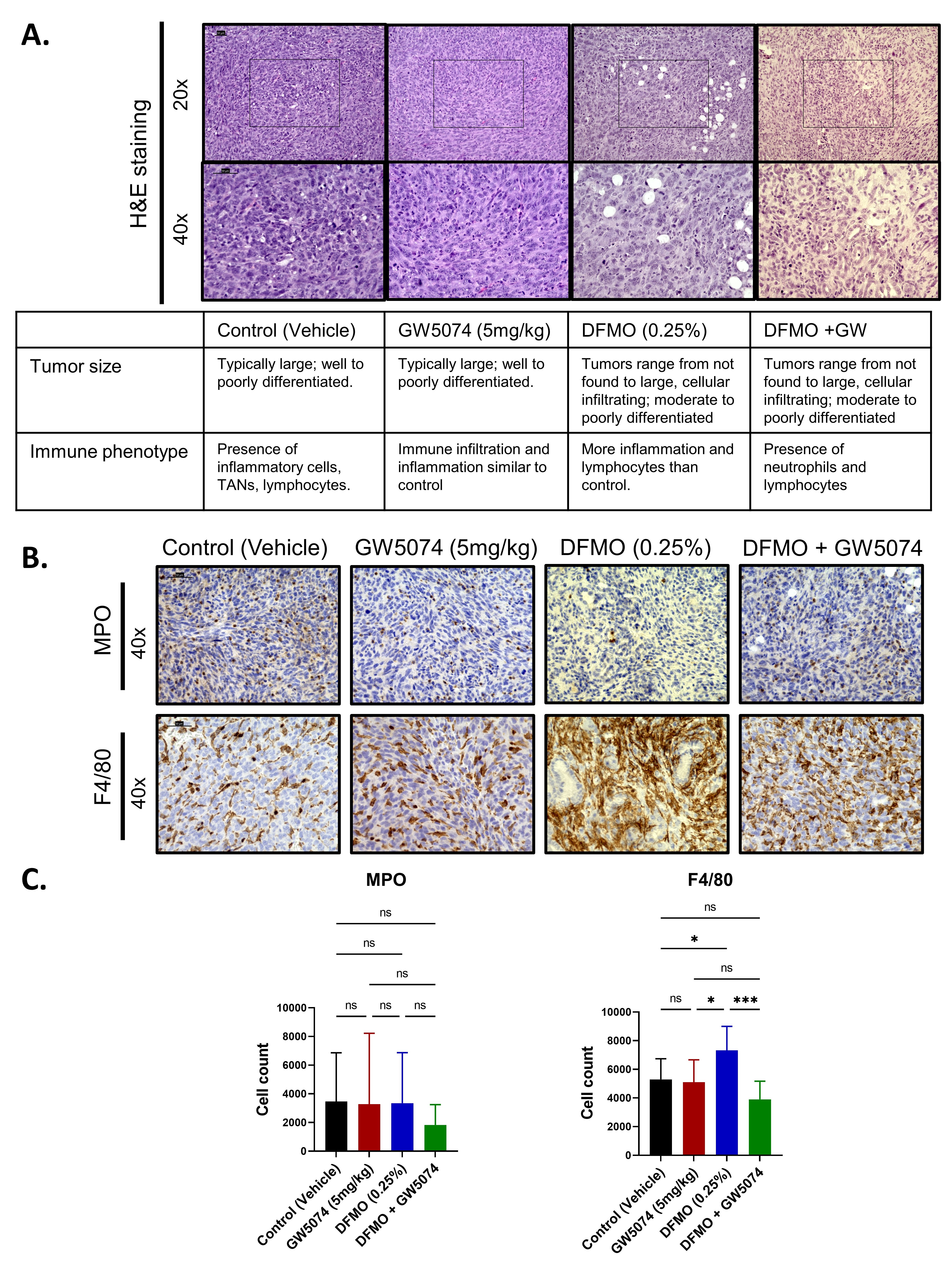
2.3. Histology Reveals Cellular Differences in DFMO Treated Pancreatic Tumors
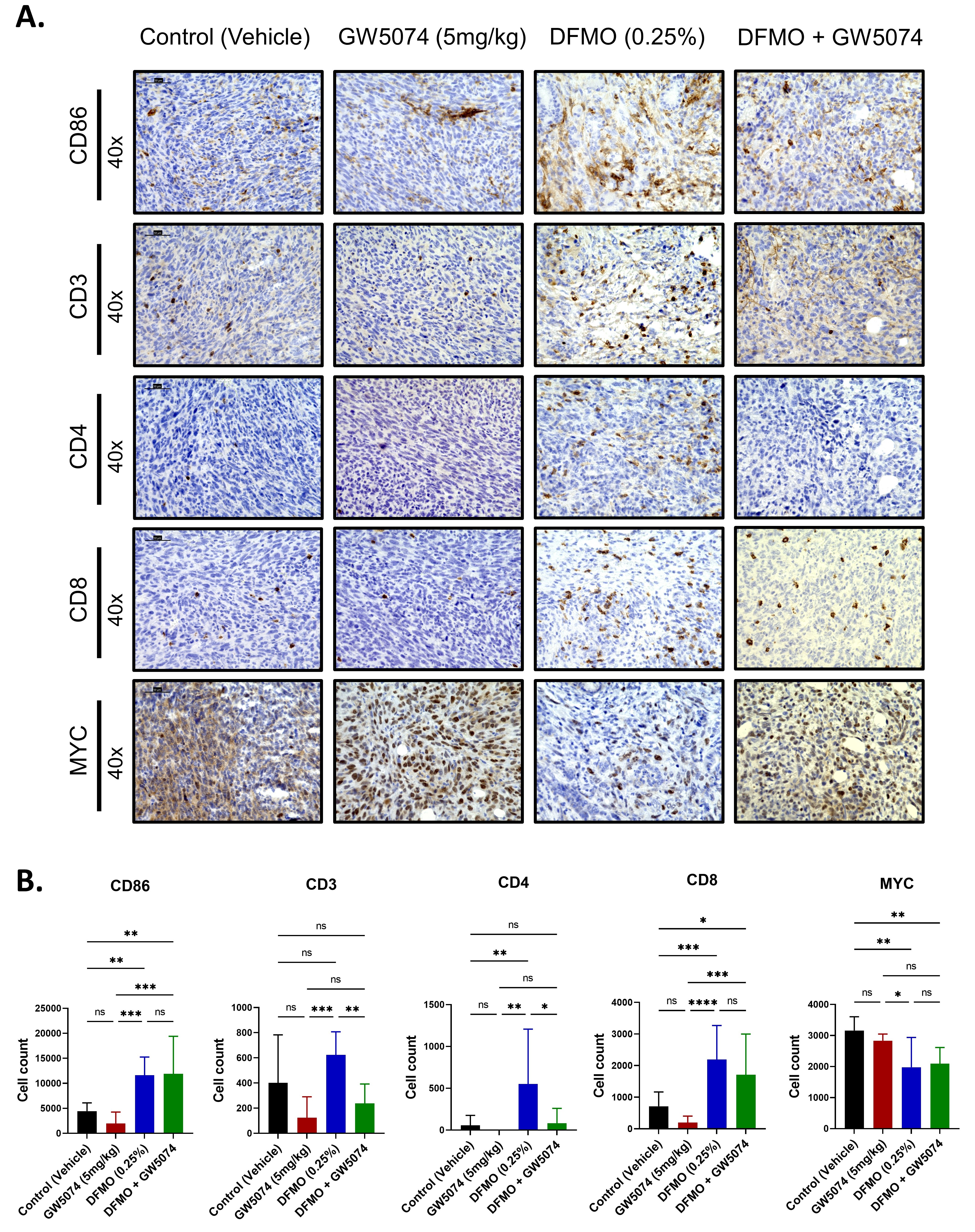
2.4. Increased T Cell Infiltration in DFMO Treated Pancreatic Tumors
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Viability
4.4. Flow Cytometry
4.5. In Vivo Testing
4.6. Histological Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Hall, B.R.; Cannon, A.; Atri, P.; Wichman, C.S.; Smith, L.M.; Ganti, A.K.; Are, C.; Sasson, A.R.; Kumar, S.; Batra, S.K. Advanced pancreatic cancer: A meta-analysis of clinical trials over thirty years. Oncotarget 2018, 9, 19396–19405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.H.; Jiang, Y.; Pillarisetty, V.G. Role of immune cells in pancreatic cancer from bench to clinical application: An updated review. Medicine 2016, 95, e5541. [Google Scholar] [CrossRef]
- Alexander, E.T.; Minton, A.; Peters, M.C.; Phanstiel, O., IV; Gilmour, S.K. A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget 2017, 8, 84140–84152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitto, S.B.; Pandey, V.; Oyer, J.L.; Copik, A.J.; Hogan, F.C.; Phanstiel, O., IV; Altomare, D.A. Difluoromethylornithine Combined with a Polyamine Transport Inhibitor Is Effective against Gemcitabine Resistant Pancreatic Cancer. Mol. Pharm. 2018, 15, 369–376. [Google Scholar] [CrossRef]
- Dobrovolskaite, A.; Madan, M.; Pandey, V.; Altomare, D.A.; Phanstiel, O., IV. The discovery of indolone GW5074 during a comprehensive search for non-polyamine-based polyamine transport inhibitors. Int. J. Biochem. Cell Biol. 2021, 138, 106038. [Google Scholar] [CrossRef]
- Chin, P.C.; Liu, L.; Morrison, B.E.; Siddiq, A.; Ratan, R.R.; Bottiglieri, T.; D’Mello, S.R. The c-Raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and Akt-independent mechanism. J. Neurochem. 2004, 90, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Pasca di Magliano, M. Kras as a key oncogene and therapeutic target in pancreatic cancer. Front. Physiol. 2013, 4, 407. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, N.; Vasseur, R.; Van Seuningen, I. The cornerstone K-RAS mutation in pancreatic adenocarcinoma: From cell signaling network, target genes, biological processes to therapeutic targeting. Crit. Rev. Oncol. Hematol. 2017, 111, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleger, C.; Verbeke, C.; Hildenbrand, R.; Zentgraf, H.; Bleyl, U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: Incidence, mechanisms, and clinical significance. Mod. Pathol. 2002, 15, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Gamble, L.D.; Purgato, S.; Murray, J.; Xiao, L.; Yu, D.M.T.; Hanssen, K.M.; Giorgi, F.M.; Carter, D.R.; Gifford, A.J.; Valli, E.; et al. Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci. Transl. Med. 2019, 11, eaau1099. [Google Scholar] [CrossRef] [Green Version]
- Sholler, G.L.S.; Ferguson, W.; Bergendahl, G.; Bond, J.P.; Neville, K.; Eslin, D.; Brown, V.; Roberts, W.; Wada, R.K.; Oesterheld, J.; et al. Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci. Rep. 2018, 8, 14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, A.; Costello, E. The role of inflammatory cells in fostering pancreatic cancer cell growth and invasion. Front. Physiol. 2012, 3, 270. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Gerner, E.W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 945–951. [Google Scholar]
- Madan, M.; Patel, A.; Skruber, K.; Geerts, D.; Altomare, D.A.; Iv, O.P. ATP13A3 and caveolin-1 as potential biomarkers for difluoromethylornithine-based therapies in pancreatic cancers. Am. J. Cancer Res. 2016, 6, 1231–1252. [Google Scholar] [PubMed]
- Muth, A.; Madan, M.; Archer, J.J.; Ocampo, N.; Rodriguez, L.; Phanstiel, O., IV. Polyamine Transport Inhibitors: Design, Synthesis, and Combination Therapies with Difluoromethylornithine. J. Med. Chem. 2014, 57, 348–363. [Google Scholar] [CrossRef]
- Geck, R.C.; Foley, J.R.; Murray Stewart, T.; Asara, J.M.; Casero, R.A.; Toker, A. Inhibition of the polyamine synthesis enzyme ornithine decarboxylase sensitizes triple-negative breast cancer cells to cytotoxic chemotherapy. J. Biol. Chem. 2020, 295, 6263–6277. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Li, Q.; Wang, J.; Pan, J.; Su, Z.; Liu, S. Dual Inhibition of Ornithine Decarboxylase and A1 Adenosine Receptor Efficiently Suppresses Breast Tumor Cells. Front. Oncol. 2021, 11, 636373. [Google Scholar] [CrossRef]
- Mohammed, A.; Janakiram, N.B.; Madka, V.; Ritchie, R.L.; Brewer, M.; Biddick, L.; Patlolla, J.M.R.; Sadeghi, M.; Lightfoot, S.; Steele, V.E.; et al. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev. Res. 2014, 7, 1198–1209. [Google Scholar] [CrossRef] [Green Version]
- Rounbehler, R.J.; Li, W.; Hall, M.A.; Yang, C.; Fallahi, M.; Cleveland, J.L. Targeting ornithine decarboxylase impairs development of MYCN-amplified neuroblastoma. Cancer Res. 2009, 69, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Bassiri, H.; Benavides, A.; Haber, M.; Gilmour, S.K.; Norris, M.D.; Hogarty, M.D. Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Transl. Pediatr. 2015, 4, 226–238. [Google Scholar] [CrossRef]
- Hong, Z.Y.; Li, S.; Liu, X.; Leng, X.M.; Miao, Z.; Kang, X.; Niu, H.; Gao, M.Q.; Lu, P. Blocking C-Raf alleviated high-dose small-volume radiation-induced epithelial mesenchymal transition in mice lung. Sci. Rep. 2020, 10, 11158. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Echeverria, V. Raf inhibitors as therapeutic agents against neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2010, 9, 120–127. [Google Scholar] [CrossRef] [PubMed]
- McCormick, F. c-Raf in KRas Mutant Cancers: A Moving Target. Cancer Cell 2018, 33, 158–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.S.K.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822.e7. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; Pasca di Magliano, M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10, 588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.-C.; Rajbhandari, N.; Liu, C.; Sakamoto, K.; Zhang, Q.; Triplett, A.A.; Batra, S.K.; Opavsky, R.; Felsher, D.W.; DiMaio, D.J.; et al. Dormant Cancer Cells Contribute to Residual Disease in a Model of Reversible Pancreatic Cancer. Cancer Res. 2013, 73, 1821. [Google Scholar] [CrossRef] [Green Version]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [Green Version]
- Casacuberta-Serra, S.; Soucek, L. Myc and Ras, the Bonnie and Clyde of immune evasion. Transl. Cancer Res. 2018, 7, S457–S459. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Hadrup, S.; Donia, M.; Thor Straten, P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron 2013, 6, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakkina, S.P.; Gitto, S.B.; Beardsley, J.M.; Pandey, V.; Rohr, M.W.; Parikh, J.G.; Phanstiel, O., IV; Altomare, D.A. DFMO Improves Survival and Increases Immune Cell Infiltration in Association with MYC Downregulation in the Pancreatic Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 13175. https://doi.org/10.3390/ijms222413175
Nakkina SP, Gitto SB, Beardsley JM, Pandey V, Rohr MW, Parikh JG, Phanstiel O IV, Altomare DA. DFMO Improves Survival and Increases Immune Cell Infiltration in Association with MYC Downregulation in the Pancreatic Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(24):13175. https://doi.org/10.3390/ijms222413175
Chicago/Turabian StyleNakkina, Sai Preethi, Sarah B. Gitto, Jordan M. Beardsley, Veethika Pandey, Michael W. Rohr, Jignesh G. Parikh, Otto Phanstiel, IV, and Deborah A. Altomare. 2021. "DFMO Improves Survival and Increases Immune Cell Infiltration in Association with MYC Downregulation in the Pancreatic Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 24: 13175. https://doi.org/10.3390/ijms222413175