
Protein Ligands in the Secretome of CD36+ Fibroblasts Induce Growth Suppression in a Subset of Breast Cancer Cell Lines

 , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Maintenance
2.2. 3D Cultures of Epithelial Cells
2.3. CD36 Transfection of Fibroblasts (FBs) and Validation
2.4. CD36 Expression in Normal Primary Fibroblasts under the Influence of Activin A
2.5. Treatment of the Tumor Cells with the CM of CD36+ FBs
2.5.1. Collecting the Conditioned Medium from FBs
2.5.2. Treatments of 3D-Cultured Epithelial Cells
2.5.3. Heat Inactivation Assay
2.5.4. Protein Fractionation of the CM of CD36+ FBs
2.5.5. Analysis of Factors Associated with Exosomes
2.6. Apoptosis Assay
2.7. Proteomics Profiling
2.7.1. Protein Digestion
2.7.2. Peptide Quantification
2.7.3. Basic Reversed-Phase Fractionation
2.7.4. Liquid Chromatography and Mass Spectrometry
2.7.5. Signal Normalization
2.8. Human Recombinant Proteins
2.9. Inhibition of KLF10 Transcription Factor
2.10. Immunofluorescence Staining
2.11. Fluorescence Microscopy
2.12. Quantification of Molecular Endpoints
2.13. Visualization of Images
2.14. Figures and Statistical Analysis
3. Results
3.1. CD36 Expression in FBs Is Reversible and Anti-Correlated with the Exposure to Activin A
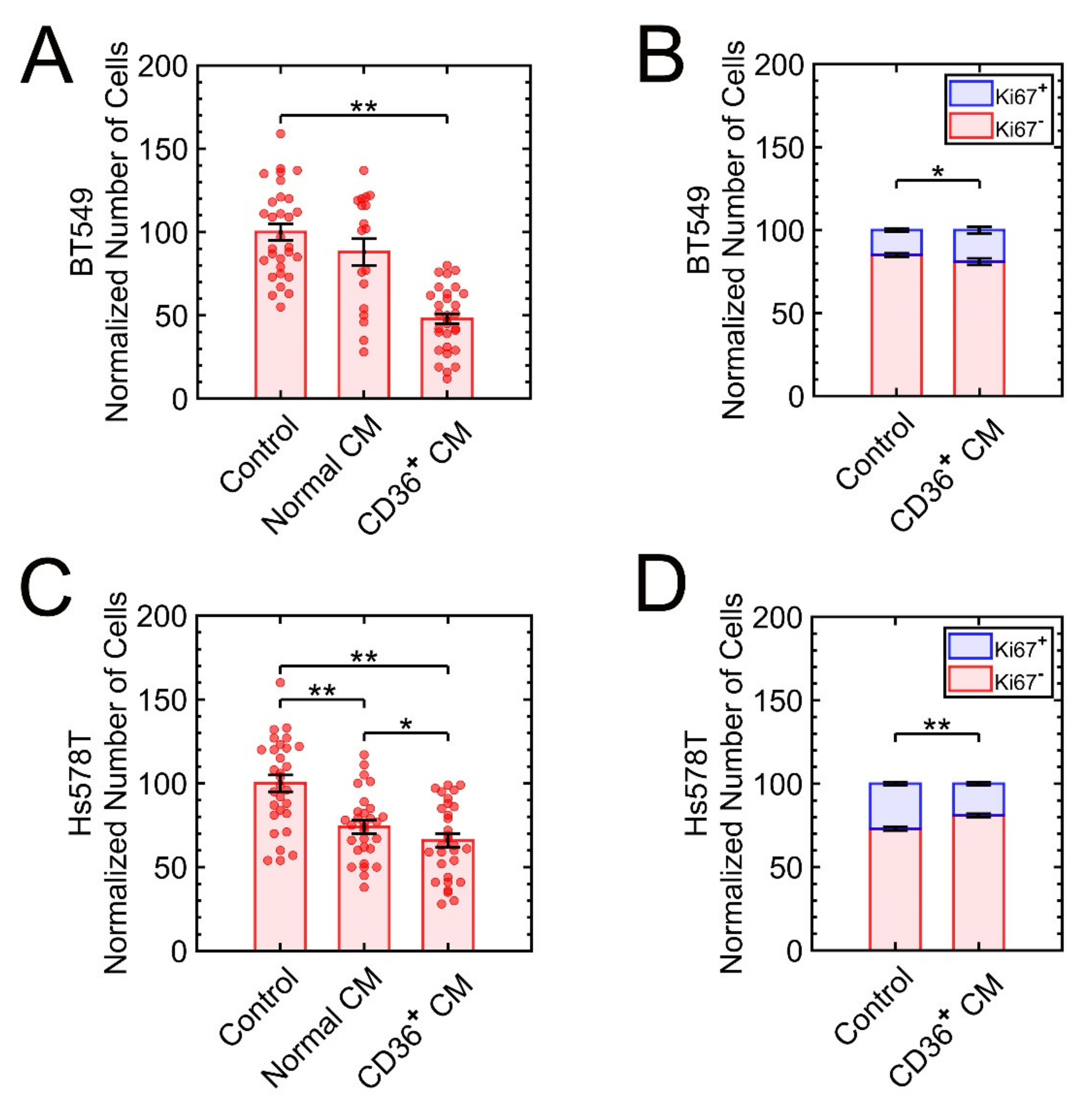
3.2. Growth Suppression by the CM of CD36+ FBs Is Subtype Dependent
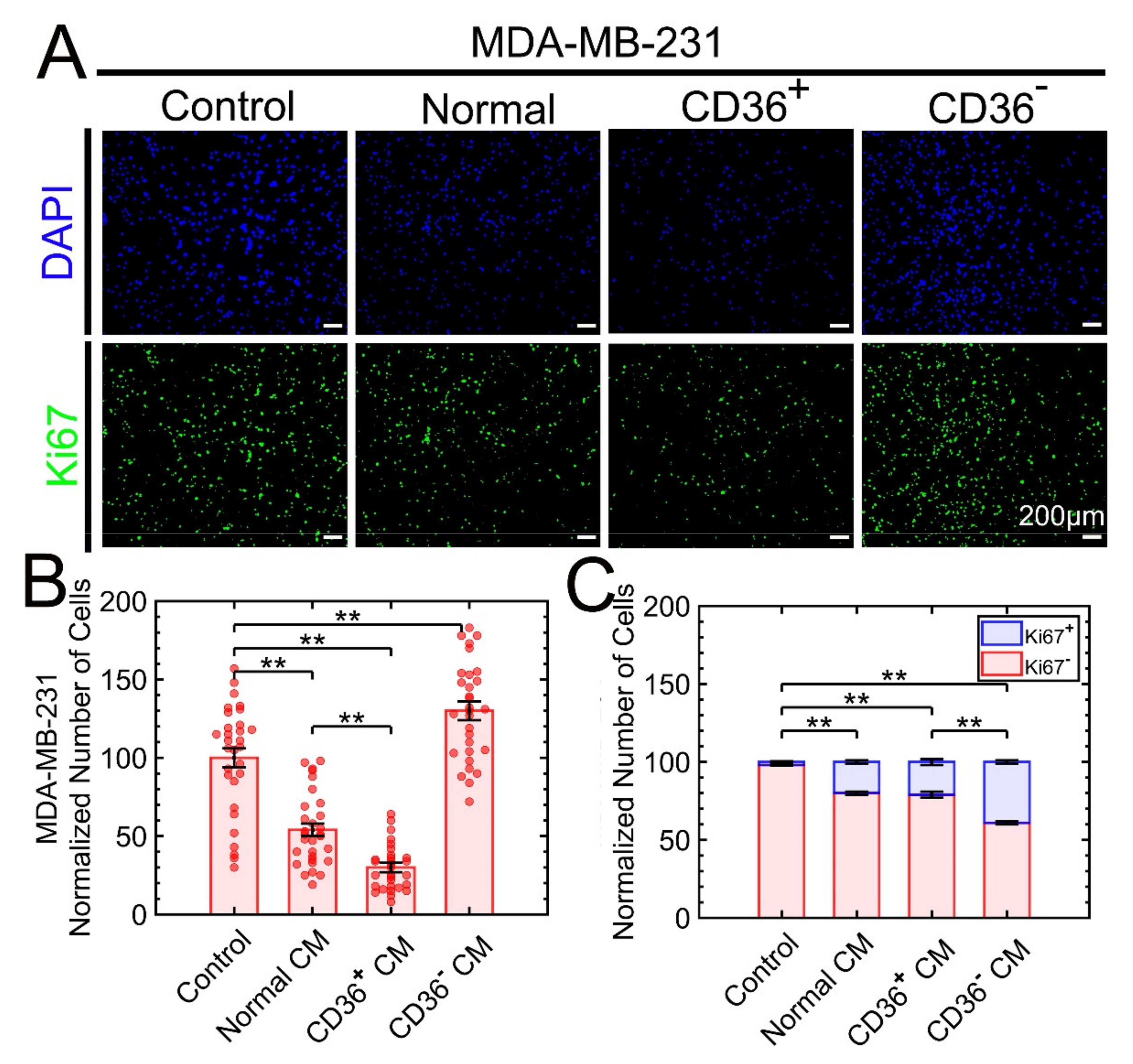
3.2.1. The Triple-Negative Breast Cancer (TNBC) Cell Lines MDA-MB-231, BT-549, and Hs578T Are Sensitive to the CM of CD36+ FBs
3.2.2. ERBB2+ Cell Line of SKBR3 Is Resistant to the CM of CD36+ FBs
3.3. MDA-MB-231 Is Used to Investigate Properties of Growth Suppression as a Function of Exposure to the CM of CD36+ FBs
3.3.1. Acute or Continuous Exposures to the CM of CD36+ FB Induce Growth Suppression
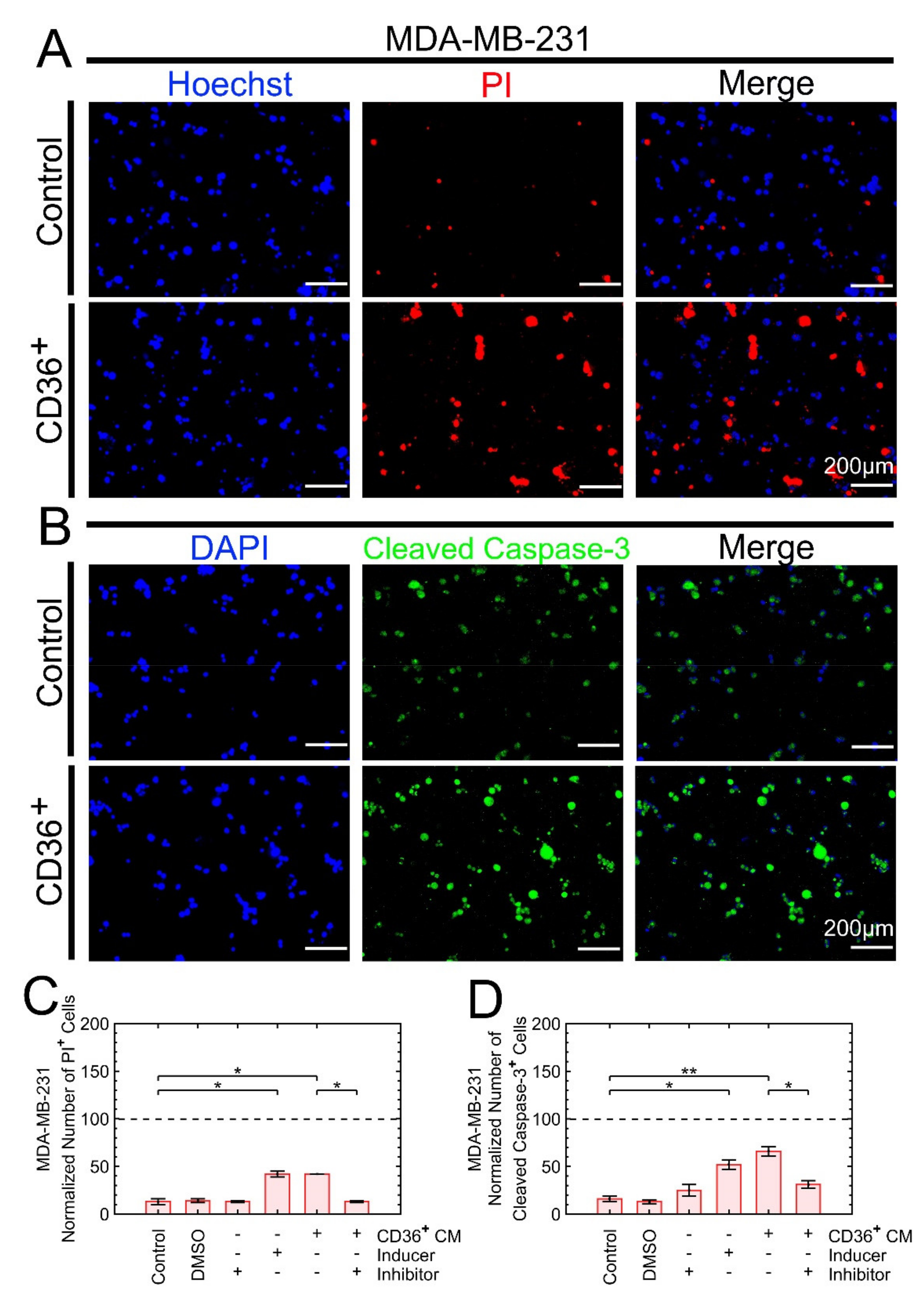
3.3.2. Growth Suppression Is Primarily due to Apoptosis
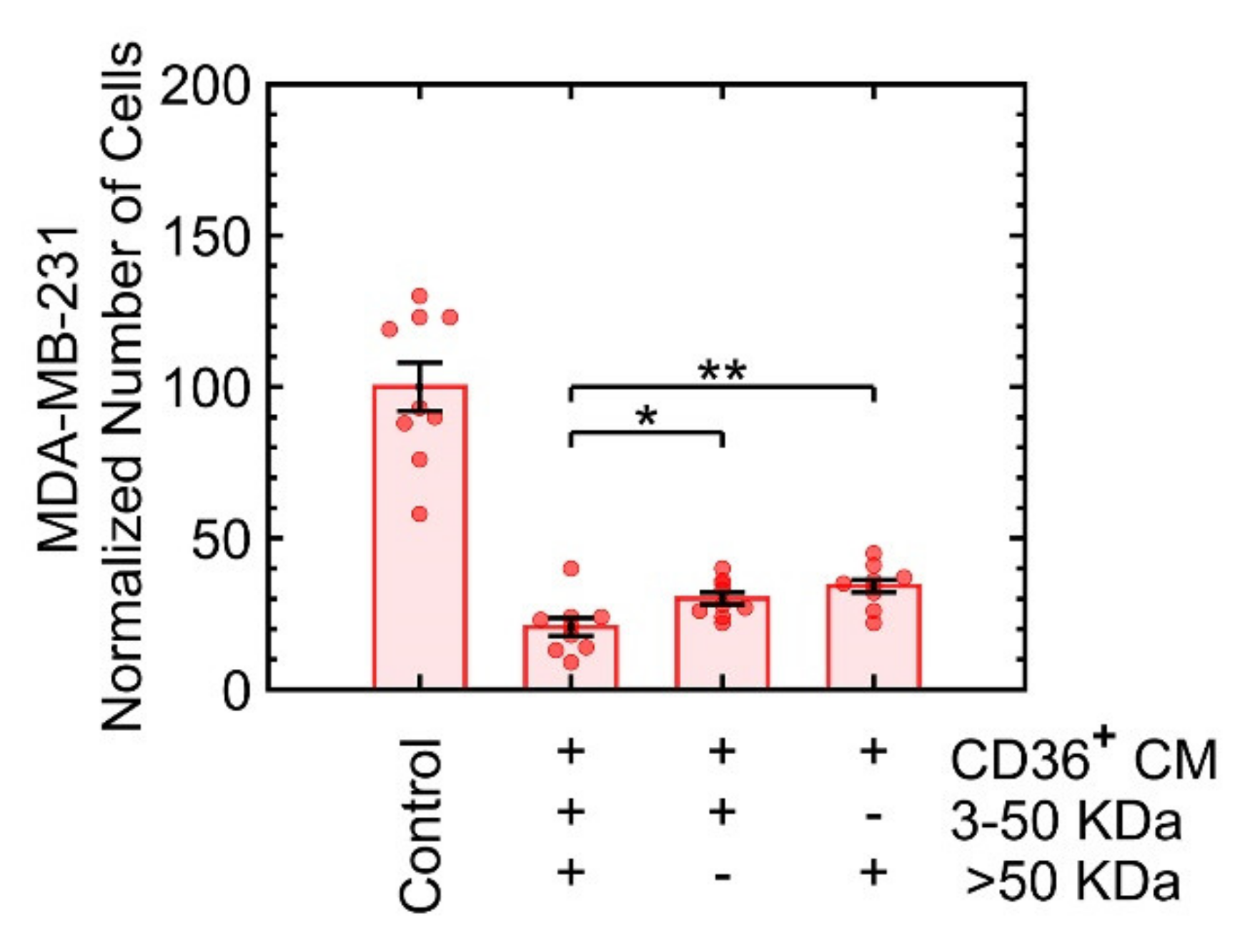
3.3.3. Growth Suppression Is Likely Induced by Proteins
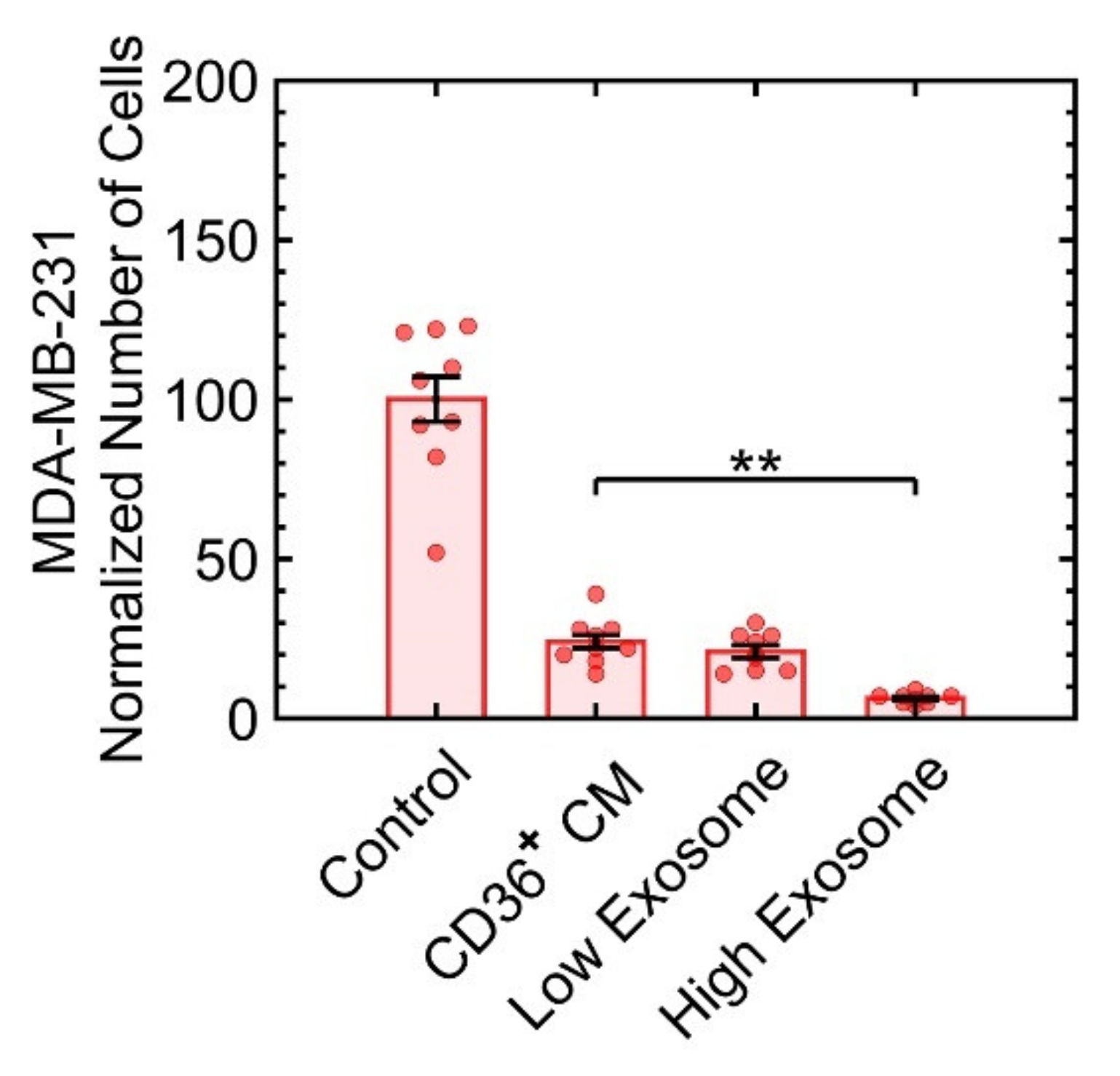
3.3.4. Exosomes Are Not the Predominant Factors in Growth Suppression
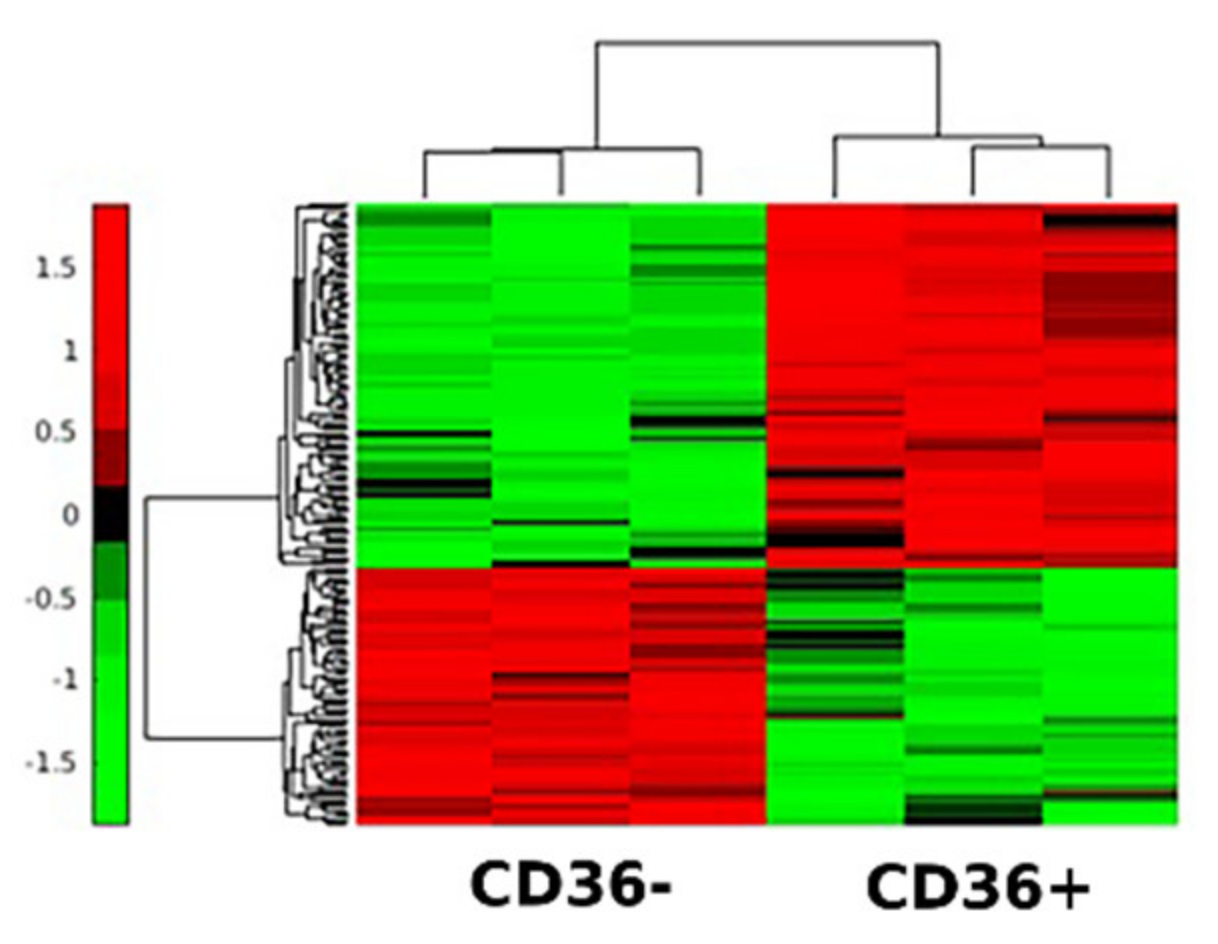
3.4. Secretomes of CD36+ versus CD36− FBs Reveal Differentially Expressed Proteins
3.5. The Ligand-Receptor Bindings Are Identified
- TIMP2 has been shown to suppress the growth and metastasis of triple-negative breast cancer in a murine model by disrupting Wnt and PI3K signaling [37].
- FBLN1, a glycoprotein member of ECM, is epigenetically downregulated in bladder cancer [40] and interacts with ADAMTS to reduce the proliferation of the breast cancer cell lines MCF7 and MDA-MB-231. ADAMTS-like protein 1 is also one of the differentially secreted proteins in the proteomics data.
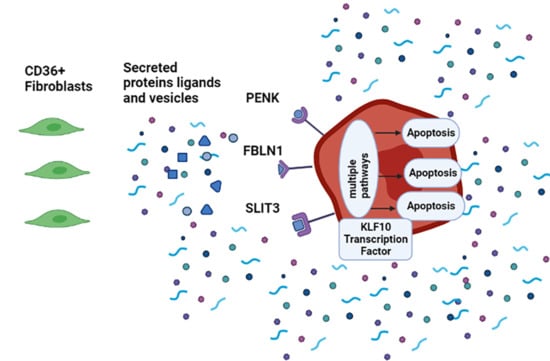
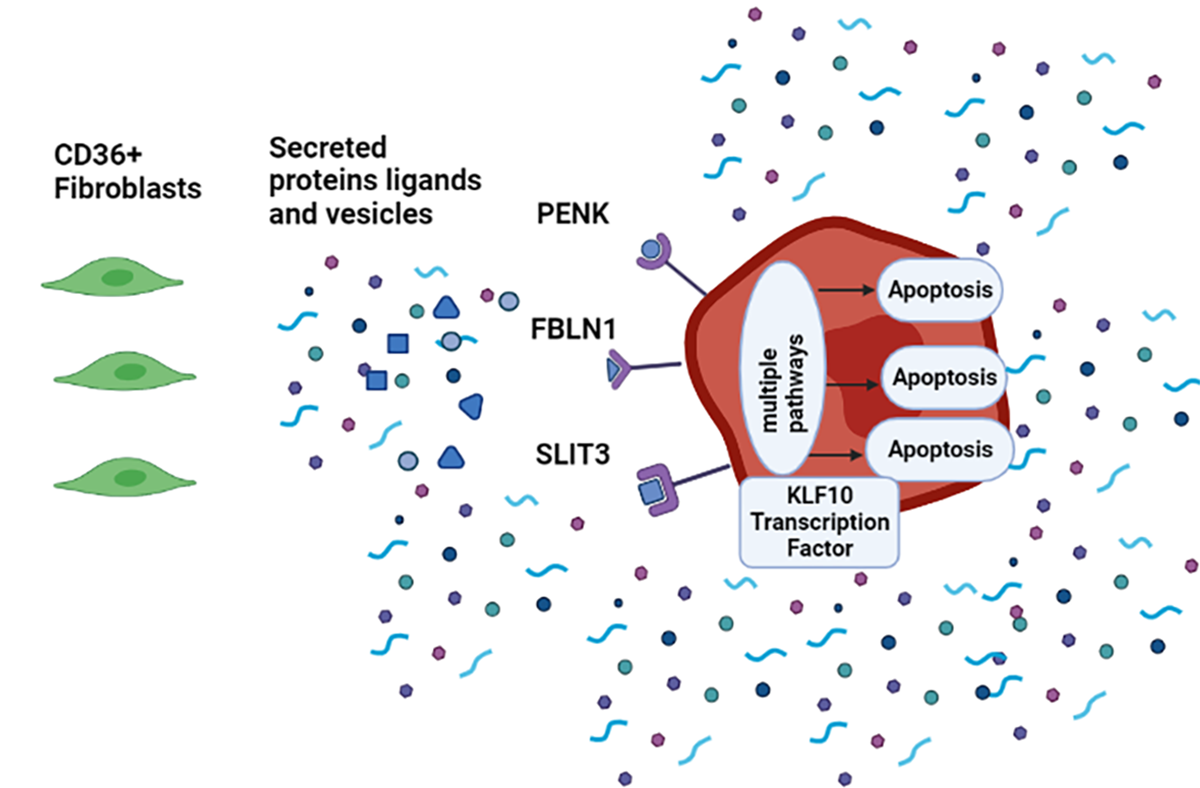
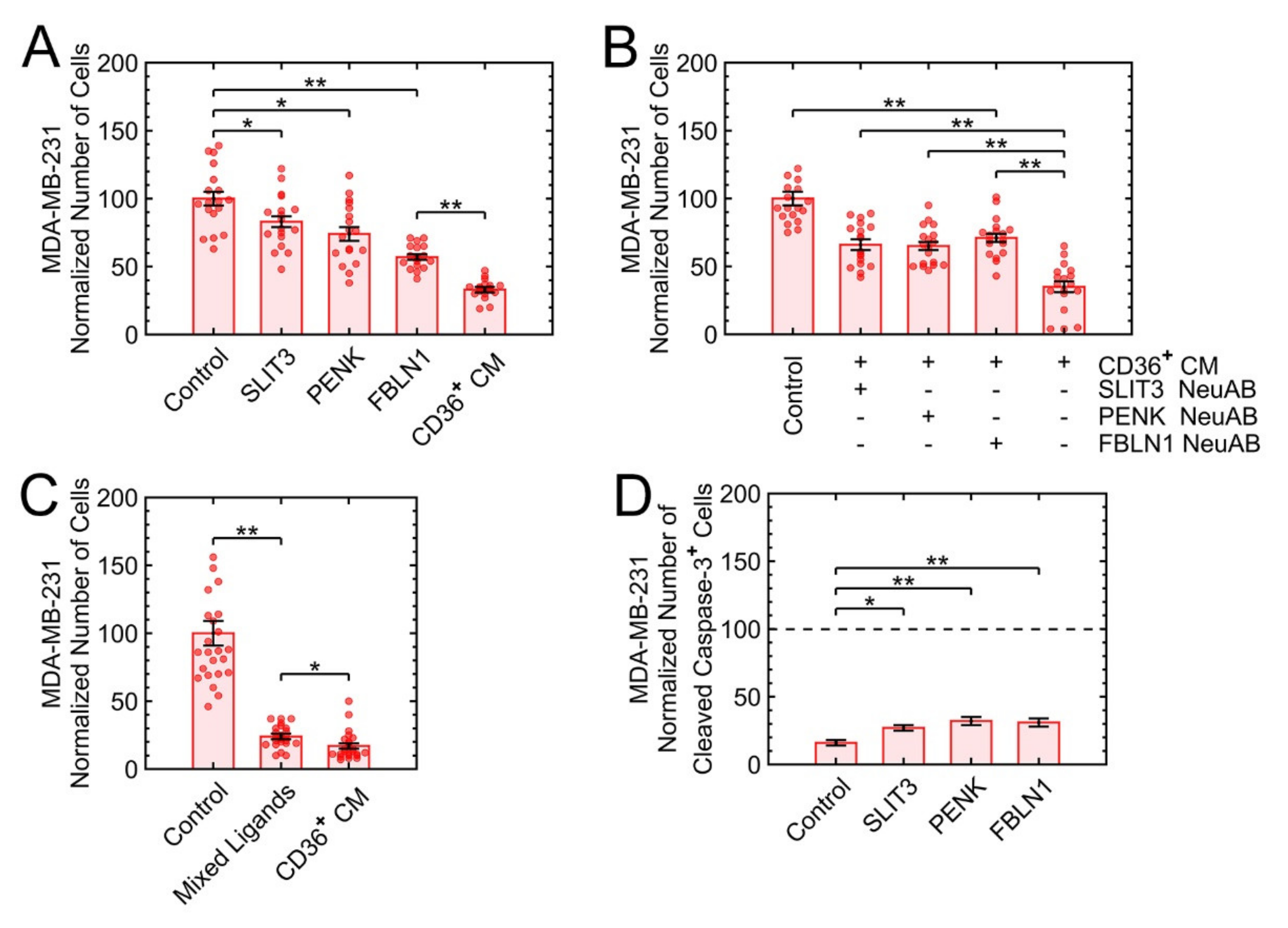
3.6. SLIT3, FBLN1, and PENK Induce Apoptosis in MDA-MB-231
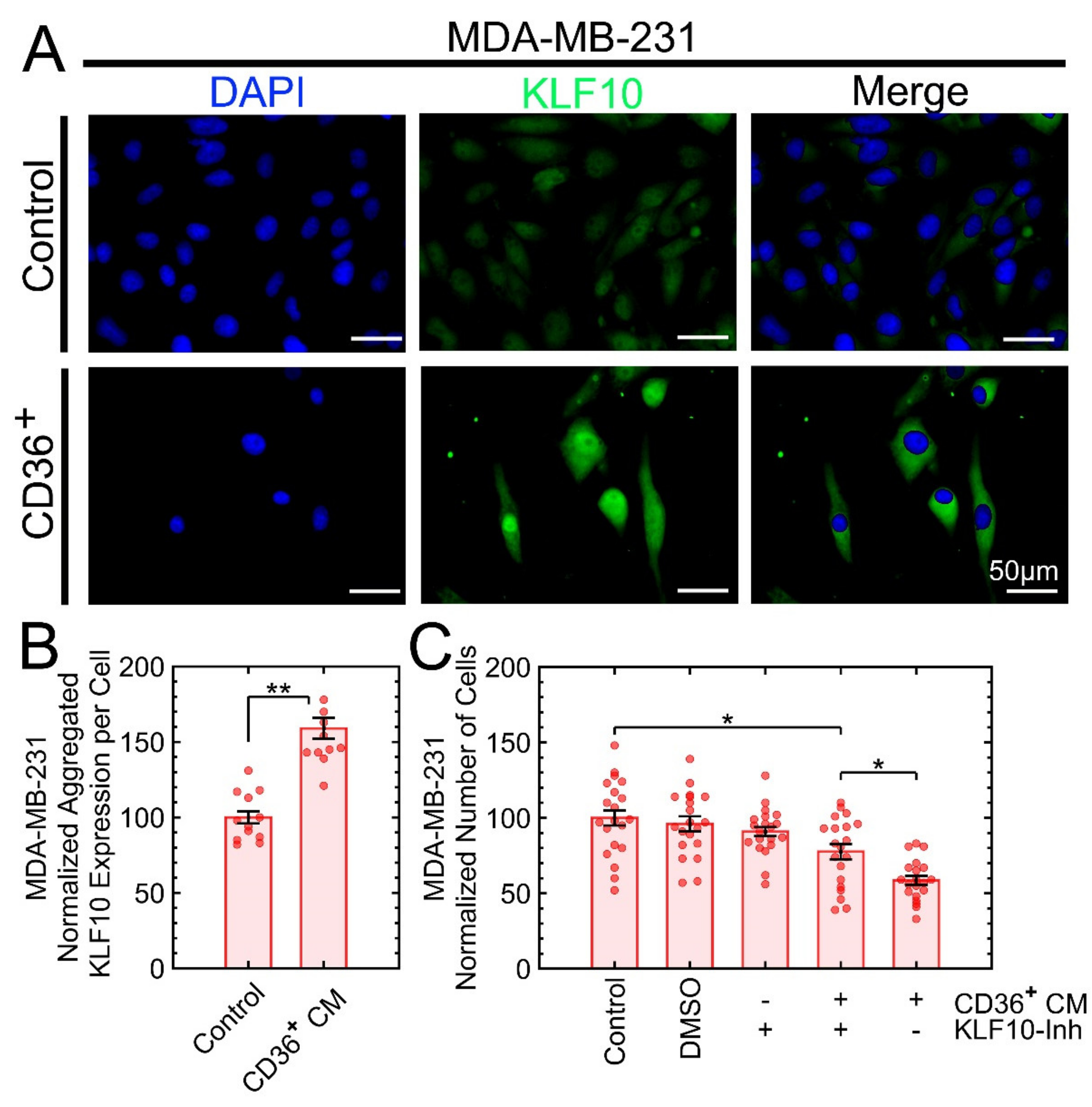
3.7. KLF10 Is One of the Transcription Factors Involved in Growth Suppression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kenny, P.A.; Bissell, M.J. Tumor reversion: Correction of malignant behavior by microenvironmental cues. Int. J. Cancer 2003, 107, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Luhr, I.; Friedl, A.; Overath, T.; Tholey, A.; Kunze, T.; Hilpert, F.; Sebens, S.; Arnold, N.; Rosel, F.; Oberg, H.H.; et al. Mammary fibroblasts regulate morphogenesis of normal and tumorigenic breast epithelial cells by mechanical and paracrine signals. Cancer Lett. 2012, 325, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.; Cukierman, E. Stromal dynamic reciprocity in cancer: Intricacies of fibroblastic-ECM interactions. Curr. Opin. Cell Biol. 2016, 42, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Giorello, M.B.; Borzone, F.R.; Labovsky, V.; Piccioni, F.V.; Chasseing, N.A. Cancer-Associated Fibroblasts in the Breast Tumor Microenvironment. J. Mammary Gland Biol. Neoplasia 2021. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Chen, Y.J.; Liao, W.X.; Huang, S.Z.; Yu, Y.F.; Wen, J.Y.; Chen, J.; Lin, D.G.; Wu, X.Y.; Jiang, N.; Li, X. Prognostic and immunological role of CD36: A pan-cancer analysis. J. Cancer 2021, 12, 4762–4773. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y. CD36 tango in cancer: Signaling pathways and functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, R.A.; Chang, H.; Dumont, N.; Rabban, J.T.; Chen, Y.Y.; Fontenay, G.V.; Berman, H.K.; Gauthier, M.L.; Zhao, J.; Hu, D.; et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012, 2, 826–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaghjyan, L.; Colditz, G.A.; Rosner, B.; Tamimi, R.M. Mammographic breast density and breast cancer risk: Interactions of percent density, absolute dense, and non-dense areas with breast cancer risk factors. Breast Cancer Res. Treat. 2015, 150, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, K.A.; Scott, C.G.; Tamimi, R.M.; Jensen, M.R.; Pankratz, V.S.; Norman, A.D.; Visscher, D.W.; Couch, F.J.; Shepherd, J.; Chen, Y.Y.; et al. Dense and nondense mammographic area and risk of breast cancer by age and tumor characteristics. Cancer Epidemiol. Biomark. Prev. 2015, 24, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, A.; Graff, R.E.; Ursin, G.; Santos Silva, I.D.; McCormack, V.; Baglietto, L.; Vachon, C.; Bakker, M.F.; Giles, G.G.; Chia, K.S.; et al. Mammographic density phenotypes and risk of breast cancer: A meta-analysis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romer, A.M.; Luhr, I.; Klein, A.; Friedl, A.; Sebens, S.; Rosel, F.; Arnold, N.; Strauss, A.; Jonat, W.; Bauer, M. Normal mammary fibroblasts induce reversion of the malignant phenotype in human primary breast cancer. Anticancer Res. 2013, 33, 1525–1536. [Google Scholar]
- Dumont, N.; Liu, B.; Defilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 2013, 15, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Khoshdeli, M.; Ferguson, B.S.; Jabbari, K.; Zang, C.; Parvin, B. YY1 is a cis-regulator in the organoid models of high mammographic density. Bioinformatics 2020, 36, 1663–1667. [Google Scholar] [CrossRef]
- Burdette, J.E.; Jeruss, J.S.; Kurley, S.J.; Lee, E.J.; Woodruff, T.K. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005, 65, 7968–7975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.; Loomans, H.A.; Le Bras, G.F.; Koumangoye, R.B.; Romero-Morales, A.I.; Quast, L.L.; Zaika, A.I.; El-Rifai, W.; Andl, T.; Andl, C.D. Activin a signaling regulates cell invasion and proliferation in esophageal adenocarcinoma. Oncotarget 2015, 6, 34228–34244. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Jabbari, K.; Winkelmaier, G.; Andersen, C.; Yaswen, P.; Khoshdeli, M.; Parvin, B. Overexpression of CD36 in mammary fibroblasts suppresses colony growth in breast cancer cell lines. Biochem. Biophys. Res. Commun. 2020, 526, 41–47. [Google Scholar] [CrossRef]
- Lee, G.Y.; Kenny, P.A.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Kim, H.; McGee, L.; Johnson, A.E.; Talwar, S.; Marugan, J.; Southall, N.; Hu, X.; Lal, M.; Mondal, D.; et al. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci. Rep. 2018, 8, 8161. [Google Scholar] [CrossRef] [Green Version]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [Green Version]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wuhr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef]
- Bilgin, C.C.; Fontenay, G.; Cheng, Q.; Chang, H.; Han, J.; Parvin, B. BioSig3D: High content screening of three-Dimensional cell culture models. PLoS ONE 2016, 11, e0148379. [Google Scholar] [CrossRef]
- Cheng, Q.; Bilgin, C.C.; Fonteney, G.; Chang, H.; Henderson, M.; Han, J.; Parvin, B. Stiffness of the microenvironment upregulates ERBB2 expression in 3D cultures of MCF10A within the range of mammographic density. Sci. Rep. 2016, 6, 28987. [Google Scholar] [CrossRef]
- Wen, Q.; Chang, H.; Parvin, B. A Delaunay triangulation approach for segmenting a clump of nuclei. In Proceedings of the IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Boston, MA, USA, 28 June–1 July 2009; pp. 9–12. [Google Scholar]
- Monti, S.; Tamayo, P.; Mesirov, J.; Golub, T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003, 52, 91–118. [Google Scholar] [CrossRef]
- Reis, F.M.; Cobellis, L.; Tameirao, L.C.; Anania, G.; Luisi, S.; Silva, I.S.; Gioffre, W.; Di Blasio, A.M.; Petraglia, F. Serum and tissue expression of activin a in postmenopausal women with breast cancer. J. Clin. Endocrinol. Metab. 2002, 87, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Veiseh, M.; Kwon, D.H.; Borowsky, A.D.; Tolg, C.; Leong, H.S.; Lewis, J.D.; Turley, E.A.; Bissell, M.J. Cellular heterogeneity profiling by hyaluronan probes reveals an invasive but slow-growing breast tumor subset. Proc. Natl. Acad. Sci. USA 2014, 111, E1731–E1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Chang, H.; Giricz, O.; Lee, G.Y.; Baehner, F.L.; Gray, J.W.; Bissell, M.J.; Kenny, P.A.; Parvin, B. Molecular predictors of 3D morphogenesis by breast cancer cell lines in 3D culture. PLoS Comput. Biol. 2010, 6, e1000684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 2006, 25, 1125–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, M.; Huang, W.; Kute, T.E.; Miller, L.D.; Zhang, Q.; Hatcher, H.; Wang, J.; Stovall, D.B.; Russell, G.B.; Cao, P.D.; et al. Yin Yang 1 plays an essential role in breast cancer and negatively regulates p27. Am. J. Pathol. 2012, 180, 2120–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenken, J.A.; Poschenrieder, A.J. Bioanalytical chemistry of cytokines--a review. Anal. Chim. Acta 2015, 853, 95–115. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. Corrigendum: A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2016, 7, 10706. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015, 6, 7866. [Google Scholar] [CrossRef] [Green Version]
- Peeney, D.; Jensen, S.M.; Castro, N.P.; Kumar, S.; Noonan, S.; Handler, C.; Kuznetsov, A.; Shih, J.; Tran, A.D.; Salomon, D.S.; et al. TIMP-2 suppresses tumor growth and metastasis in murine model of triple-negative breast cancer. Carcinogenesis 2020, 41, 313–325. [Google Scholar] [CrossRef]
- Jiang, Z.; Liang, G.; Xiao, Y.; Qin, T.; Chen, X.; Wu, E.; Ma, Q.; Wang, Z. Targeting the SLIT/ROBO pathway in tumor progression: Molecular mechanisms and therapeutic perspectives. Adv. Med. Oncol. 2019, 11, 1758835919855238. [Google Scholar] [CrossRef]
- Marlow, R.; Strickland, P.; Lee, J.S.; Wu, X.; Pebenito, M.; Binnewies, M.; Le, E.K.; Moran, A.; Macias, H.; Cardiff, R.D.; et al. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res. 2008, 68, 7819–7827. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, J.; Li, H.; Xia, D.; Yu, G.; Yao, W.; Yang, Y.; Xiao, H.; Lang, B.; Ma, X.; et al. Fibulin-1 is epigenetically down-regulated and related with bladder cancer recurrence. BMC Cancer 2014, 14, 677. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.L.; Liu, W.J.; Chen, Q.; Liu, J.; Yang, C.Q.; Zhang, G.; Zhang, S.L.; Guo, W.H.; Li, J.B.; Zhao, G.; et al. miR-506-loaded gelatin nanospheres target PENK and inactivate the ERK/Fos signaling pathway to suppress triple-negative breast cancer aggressiveness. Mol. Carcinog. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.F.; Lin, T.L.; Wang, Y.Y.; Cao, H. High expression of proenkephalin is associated with favorable outcomes in patients with gastrointestinal stromal tumors. Cancer Manag. Res. 2019, 11, 6681–6690. [Google Scholar] [CrossRef] [Green Version]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borras, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A Compendium for Extracellular Vesicles with Continuous Community Annotation. PLoS Biol. 2012, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geutskens, S.B.; Hordijk, P.L.; van Hennik, P.B. The chemorepellent Slit3 promotes monocyte migration. J. Immunol. 2010, 185, 7691–7698. [Google Scholar] [CrossRef]
- Twal, W.O.; Czirok, A.; Hegedus, B.; Knaak, C.; Chintalapudi, M.R.; Okagawa, H.; Sugi, Y.; Argraves, W.S. Fibulin-1 suppression of fibronectin-regulated cell adhesion and motility. J. Cell Sci. 2001, 114, 4587–4598. [Google Scholar] [CrossRef]
- Tran, H.; Tanaka, A.; Litvinovich, S.V.; Medved, L.V.; Haudenschild, C.C.; Argraves, W.S. The interaction of fibulin-1 with fibrinogen. A potential role in hemostasis and thrombosis. J. Biol. Chem. 1995, 270, 19458–19464. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Civelek, M.; Miller, C.L.; Sheffield, N.C.; Guertin, M.J.; Zang, C. BART: A transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics 2018, 34, 2867–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, G.; Feng, L.; Lu, H.; Wang, X. Kruppel-like factors in breast cancer: Function, regulation and clinical relevance. Biomed. Pharm. 2020, 123, 109778. [Google Scholar] [CrossRef]
- Tetreault, M.P.; Yang, Y.; Katz, J.P. Kruppel-like factors in cancer. Nat. Rev. Cancer 2013, 13, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Yoda, T.; McNamara, K.M.; Miki, Y.; Onodera, Y.; Takagi, K.; Nakamura, Y.; Ishida, T.; Suzuki, T.; Ohuchi, N.; Sasano, H. KLF15 in breast cancer: A novel tumor suppressor? Cell. Oncol. Dordr. 2015, 38, 227–235. [Google Scholar] [CrossRef]
- Spittau, B.; Krieglstein, K. Klf10 and Klf11 as mediators of TGF-beta superfamily signaling. Cell Tissue Res. 2012, 347, 65–72. [Google Scholar] [CrossRef]
- Memon, A.; Lee, W.K. KLF10 as a Tumor Suppressor Gene and Its TGF-beta Signaling. Cancers 2018, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, R.E.; Dallol, A.; Bieche, I.; Krex, D.; Morton, D.; Maher, E.R.; Latif, F. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br. J. Cancer 2004, 91, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Guo, H.; Li, B.; Sui, C.; Zhang, Y.; Xia, X.; Qin, Y.; Ye, L.; Xie, F.; Wang, H.; et al. Effects of Slit3 silencing on the invasive ability of lung carcinoma A549 cells. Oncol. Rep. 2015, 34, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C. Agonists and Antagonists of TGF-beta Family Ligands. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harikrishnan, K.; Joshi, O.; Madangirikar, S.; Balasubramanian, N. Cell Derived Matrix Fibulin-1 Associates With Epidermal Growth Factor Receptor to Inhibit Its Activation, Localization and Function in Lung Cancer Calu-1 Cells. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Jie, Y.S.; Xiao, C.C.; Zhou, W.Y.; Li, X.H.; Chen, Y.Q.; Wu, Y.K.; Cao, J.; Zhang, Q.; Gao, Z.L.; et al. Increased Expression of Fibulin-1 Is Associated With Hepatocellular Carcinoma Progression by Regulating the Notch Signaling Pathway. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Sadlonova, A.; Bowe, D.B.; Novak, Z.; Mukherjee, S.; Duncan, V.E.; Page, G.P.; Frost, A.R. Identification of molecular distinctions between normal breast-associated fibroblasts and breast cancer-associated fibroblasts. Cancer Microenviron. 2009, 2, 9–21. [Google Scholar] [CrossRef] [Green Version]
- McTavish, N.; Copeland, L.A.; Saville, M.K.; Perkins, N.D.; Spruce, B.A. Proenkephalin assists stress-activated apoptosis through transcriptional repression of NF-kappa B- and p53-regulated gene targets. Cell Death Differ. 2007, 14, 1700–1710. [Google Scholar] [CrossRef]
- Yang, H.; Li, Q.; Wang, C.; Wang, J.; Lv, J.; Wang, L.; Zhang, Z.S.; Yao, Z.; Wang, Q. Cytotoxic Necrotizing Factor 1 Downregulates CD36 Transcription in Macrophages to Induce Inflammation During Acute Urinary Tract Infections. Front. Immunol. 2018, 9, 1987. [Google Scholar] [CrossRef]
- Pupa, S.M.; Argraves, S.W.; Forti, S.; Casalini, P.; Berno, V.; Agresti, R.; Aiello, P.; Invernizzi, A.; Baldassari, P.; Otwal, W.; et al. Immunological and pathobiological roles of fibulin-1 in breast cancer (vol 23, pg 2153, 2004). Oncogene 2004, 23, 6325. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | CD36 | YY1 | Ki67 | KLF10 | Cleaved Caspase-3 | Cyclin E1 |
|---|---|---|---|---|---|---|
| Permeabilization (Triton X-100) | 0.5% | 0.5% | 0.5% | 0.5% | 0.3% | 0.5% |
| Blocking Solution (BSA) | 1% | 1% | 1% | 1% | 5% BSA + 0.3% Triton X-100 | 1% |
| Primary Antibody | Santa Cruz SC-7309 Or Novus Biological NB 400-144 | Abcam Ab232573 | Abcam Ab16667 | Novus Biological NBP3-04586 | Abcam Ab32042 | Novus Biological NBP2-67760 |
| 1:250 | 1:250 | 1:500 | 1:100 | 1:100 | 1:100 | |
| Secondary Antibody | Abcam Ab150113 Or Ab150077 | Abcam Ab175471 | Abcam Ab150077 | Abcam Ab150077 | Abcam Ab150077 | Abcam Ab150077 |
| 1:250 | 1:250 | 1:500 | 1:250 | 1:250 | 1:250 |
| Ligand | Receptors | p-Value | Log2 (Fold Change) |
|---|---|---|---|
| TIMP2 (24 KDa) | ITGA3, ITGB1 | 0.00005 | 0.7 |
| FBLN1 (77 KDa) | ITGB1 | 0.001 | 0.5 |
| PENK (31 KDa) | MRGPRX1, | 0.001 | 0.5 |
| OGFR, | |||
| OPRD1, | |||
| OPK1, | |||
| OPRM1 | |||
| SLIT3 (168 KDa) | ROBO2 | 0.03 | 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabbari, K.; Winkelmaier, G.; Andersen, C.; Yaswen, P.; Quilici, D.; Furuta, S.; Cheng, Q.; Parvin, B. Protein Ligands in the Secretome of CD36+ Fibroblasts Induce Growth Suppression in a Subset of Breast Cancer Cell Lines. Cancers 2021, 13, 4521. https://doi.org/10.3390/cancers13184521
Jabbari K, Winkelmaier G, Andersen C, Yaswen P, Quilici D, Furuta S, Cheng Q, Parvin B. Protein Ligands in the Secretome of CD36+ Fibroblasts Induce Growth Suppression in a Subset of Breast Cancer Cell Lines. Cancers. 2021; 13(18):4521. https://doi.org/10.3390/cancers13184521
Chicago/Turabian StyleJabbari, Kosar, Garrett Winkelmaier, Cody Andersen, Paul Yaswen, David Quilici, Saori Furuta, Qingsu Cheng, and Bahram Parvin. 2021. "Protein Ligands in the Secretome of CD36+ Fibroblasts Induce Growth Suppression in a Subset of Breast Cancer Cell Lines" Cancers 13, no. 18: 4521. https://doi.org/10.3390/cancers13184521