
Abstract
Biomedical research is undergoing a paradigm shift towards approaches centred on human disease models owing to the notoriously high failure rates of the current drug development process. Major drivers for this transition are the limitations of animal models, which, despite remaining the gold standard in basic and preclinical research, suffer from interspecies differences and poor prediction of human physiological and pathological conditions. To bridge this translational gap, bioengineered human disease models with high clinical mimicry are being developed. In this Review, we discuss preclinical and clinical studies that benefited from these models, focusing on organoids, bioengineered tissue models and organs-on-chips. Furthermore, we provide a high-level design framework to facilitate clinical translation and accelerate drug development using bioengineered human disease models.
Key points
-
Advances in bioengineering have yielded complex human disease models with high clinical biomimicry and predictability.
-
Human disease models can help unravel disease mechanisms, including for infectious and genetic diseases and cancer.
-
Using appropriate human disease models in the drug development process and clinical decision-making improves the rate of clinical translation, reduces costs and directly benefits patients.
-
Stringent model validation, regulatory and legal guidance, and scalable disease model production are key future milestones to facilitate their implementation in (pre-)clinical research.
Similar content being viewed by others

Introduction
Biomedical research is currently undergoing a paradigm shift towards approaches centred on human disease models1. This shift has been driven by the notoriously high failure rates of the current drug development process. Although investments increased at unprecedented rates over the past decade (US$133 billion research and development expenditures of the 15 biggest pharma companies in 2021, a 44% increase since 2016)2, the drug attrition rate hit an all-time high of 95% in 2021 (ref. 3). Most drugs fail in clinical stages (Fig. 1) despite proven efficacy and safety in animal models4,5. Different reasons account for this translational gap, one of them being that the decision on entry of a drug candidate into clinical trials relies almost exclusively on animal-derived data.

Current development pipeline of new drugs with proven preclinical safety and efficacy in animal models. The average duration of the different (pre-)clinical stages, the percentage of drugs (averaged over the past 5 years) that move to the next phase and the median costs of the different stages per drug are illustrated3,180.
Animal models, however, often fail to filter out harmful or ineffective drugs6. Moreover, potentially effective drug candidates never enter clinical trials owing to negative preclinical tests given that most animal models poorly resemble human conditions and thus have low predictive values. The discrepancies derive from different anatomical layouts and biological barriers, divergent receptor expression and immune responses, host specificities of microorganisms, and distinct pathomechanisms. In addition, animals are inbred and kept under standardized conditions and thus do not account for the genetic and ethnic diversity of humans. Therefore, drug safety or efficacy issues that only affect certain subpopulations go unnoticed.
Furthermore, fast-paced advances in genome editing and antibody therapies have direct implications for the drug development process. Currently, 40% of the drugs undergoing clinical trials are antibodies3. However, their high target specificity requires the identification of cross-reactive species during preclinical testing. Non-human primates are often the only pharmacologically relevant species, whose use has ethical and economic implications7. Species-specificity is also a concern for gene therapies because genetic sequences and therapeutic efficacy differ between animals. For example, base editors yield 61% in vivo gene editing efficacy in the liver of mice compared to 26% in primates8.
The COVID-19 pandemic further highlighted the model dilemma in biomedical research. At the beginning of the pandemic, it was unclear which species were susceptible to SARS-CoV-2 and no model was readily available to study the course of the disease and identify druggable targets against the unknown pathogen9. Suitable and readily available disease models could have substantially accelerated our understanding of virus–host interactions and expedited the identification of effective drugs in repurposing studies.
In view of the deficits of such an animal-centred system, efforts are ongoing to develop bioengineered human-based (disease) models of high clinical biomimicry to close this translational gap.
This Review discusses success stories and applications of human (disease) models in preclinical and clinical research, focusing on organoids, bioengineered tissue models and organs-on-chips (OoCs).
Overview of human disease models
Different disease models are available covering a broad range of physiological and pathological conditions. An overview of their characteristics, advantages and disadvantages is provided herein.
2D cell cultures
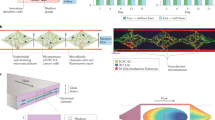
Two-dimensional (2D) cultivated patient-derived cells are an invaluable tool to study disease phenotypes and pathomechanisms, especially during the early phases of drug development (Fig. 2). Primary cells are the preferred option owing to their higher genetic heterogeneity compared to cell lines, but their limited availability or in vitro proliferation capacity restricts their use. Multipotent adult stem cells (ASCs) and induced pluripotent stem (iPS) cells can help overcome this shortage, as they can readily and indefinitely propagate and convert into any somatic cell10. Engineered cells, such as reporter cell lines, are also commonly used as they are amenable to high-throughput manufacturing and have high reproducibility and lower costs relative to stem cell-based methods.

From left to right, bioengineered models are ordered from the least to the most complex, including their advantages, limitations and stage of application during the drug development process. iPS cell, induced pluripotent stem cell.
Nonetheless, 2D cell cultures have limitations. For example, the epithelial differentiation stages of stratified epithelia, such as the skin, cannot be mimicked in 2D. Moreover, cell responses and gene and/or protein expression patterns greatly differ between 2D and three-dimensional (3D) models11. For example, 3D cultures of lung fibroblasts resemble in vivo tumour necrosis factor (TNF) receptor expression and nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) activation patterns more closely than 2D cultures12. Similarly, the biomimicry of tissue-specific transporters and cell junctions is often higher in 3D models13.
Bioengineered tissue models
Bioengineered tissue models are primarily generated from human stem cells or primary cells, the latter of which are isolated from surgically excised human tissue or from non-transplantable organs (Fig. 2). The cells are manually added or 3D bioprinted onto or into hydrogel or polymer-based scaffolds14, often in transwell setups15. Alternatively, de-cellularized extracellular matrix scaffolds from animal-derived organs16 or non-transplantable human organs17 can be reseeded with human cells. The advantages of these models include the possibility to cultivate them at the air–liquid interface or submerged, thereby emulating in vivo-like tissue conditions and their maturated and differentiated tissue state, and achieving increased biomimicry of native human tissue compared to 2D18,19. This approach is often used for multi-layer or stratified tissues such as the gut, lungs and skin20,21 but also the brain and liver as it allows a controlled build-up of the model22,23. However, these tissue models cannot be cryopreserved or propagated and therefore have a limited lifespan. Furthermore, they have limited cell diversity and lack self-renewal capacity in non-stem cell-based models.
Organoids
Organoids are self-organizing 3D structures generated from tissue-specific ASCs or iPS cells24 (Fig. 2). Stem cells are embedded in an extracellular matrix that provides the scaffold for tissue growth. Organoid formation is then guided by a cocktail of growth factors that are pivotal for tissue development in vivo, which enables stem cells to maintain their differentiation and self-renewal capacity. The size of these organ-like structures ranges from 100 µm (lung organoid) to 2 mm (brain organoid) and they can be cultivated in small dimensions (for example, 96-well or 384-well plates25,26), thereby being suitable for high-throughput screening.
Protocols for the generation of iPS cell-derived organoids are available for a variety of human organs, including the gut27, stomach28, liver29, pancreas30, lung31, thyroid32, kidney33, retinal and optic cup34, and brain35. However, iPS cell-derived organoids fail to mature beyond the fetal phenotype unless they are grafted into living organisms36,37. To overcome this limitation, the use of tissue-resident ASCs derived from postnatal or adult tissues has gained increasing attention, resulting in organoids of endoderm-derived tissues such as lung38, gut39, pancreas40, stomach41, liver organoids42, endometrium43, prostate44, fallopian tube45 and mammary gland46. Although the application of ASCs has been hampered by their limited availability in the past, some commercial suppliers now provide ASC-derived organoids. Nonetheless, ASC-derived organoids lack cells from the ectodermal and mesenchymal germ layer and are usually smaller than iPS cell-derived counterparts. Furthermore, the cell type composition of organoids of the same organ can vary depending on the protocol47, ultimately hindering reproducibility.
Organs-on-chips
Inter-tissue crosstalk and complex in vivo-like processes cannot be mimicked in single and static tissue models. To enable tissue perfusion and dynamics as well as multi-organ crosstalk, OoCs have been introduced48,49,50,51 (Fig. 2). OoCs are perfused microfluidic platforms that contain bioengineered or miniaturized tissues or organs interconnected by 3D microchannels to simulate the in vivo functions, biomechanics and (patho)physiological responses of organs6,52,53,54. OoC setups are often referred to as microphysiological systems, as they can emulate human (patho)physiology in a more human-like environment49,51,55,56,57. As opposed to organoids that form by self-organization, OoCs follow a reductionist engineering approach through the targeted and pre-defined design of components, such as the scaling of cell numbers used based on their physiological function, or including disease-relevant cell types and key biophysical and biochemical cues24.
OoCs can be single-organ or multi-organ systems. Single-organ systems can assess the response of a specific tissue or organ to a particular stimulus. By contrast, multi-organ systems allow study of the communication and interactions of several tissues and organs simultaneously. Notably, the interconnection of at least 10 human organs, including circulatory, endocrine, gastrointestinal, immune, integumentary, musculoskeletal, nervous, reproductive, respiratory and urinary, in OoCs has been hypothesized to provide sufficient complexity to resemble a ‘human-on-a-chip’58,59,60. Furthermore, biophysical processes, such as chip perfusion, can be easily automated using OoCs, which allows monitoring of tissue function and responses in situ and in real time52. For these reasons, OoCs currently constitute the most promising approach to emulate human diseases in vitro55,61. However, they are still complex and thereby not amenable to high-throughput methods.
Preclinical and clinical applications
Preclinical research
Human-based models are invaluable tools in every stage of the drug development process, from high-throughput screens to identify target or lead molecules, to efficacy and safety testing in preclinical stages and implementation in clinical trials and decision-making. Nonetheless, human-based models have so far been primarily used for toxicity testing61,62,63,64, and their application in preclinical and clinical research is still in its infancy. Promising disease models of the liver65,66, pancreas67,68, central nervous system35,69,70, skin71, lung72,73, intestine72,73,74,75,76, musculoskeletal system77 and heart78,79,80,81 have been developed, but their equivalence or superiority over their animal models needs to be demonstrated. In this section, successful examples of preclinical and clinical implementation of human-based disease models are discussed.
Unravelling disease mechanisms and target identification
Rational drug design and development require a proper understanding of the underlying disease mechanism. Human disease models can facilitate the process as evidenced by ad hoc systems that emerged during the COVID-19 pandemic1. For example, central pathomechanisms, such as the contribution of endothelial dysfunction, the ‘cytokine storm’ and the intercellular variability of infection susceptibility, were identified in organoids and lung-on-a-chip models82,83,84,85,86. A vasculature-on-a-chip setup revealed that SARS-CoV-2 exposure substantially reduces endothelial barrier function by perturbing vascular endothelial–cadherin junctions and increasing pro-inflammatory cytokine release. This endothelial dysfunction is exacerbated after the introduction of peripheral blood mononuclear cells, resulting in excessive inflammation82. Organoids demonstrated the induction of inefficient interferon responses in SARS-CoV-2 infections84,85, revealing similar interferon signatures between SARS-CoV-2-infected bioengineered lung models and native human lungs, highlighting the value and predictivity of human models84.
Bioengineered tissues also contributed to the understanding of the effects of COVID-19 beyond the lungs. For example, SARS-CoV-2 tropism for neurons87 and cortical astrocytes88 or its preference for mature cell types and distinct neurotoxic effects were demonstrated in brain organoids. Interestingly, the expression levels of the SARS-CoV-2 receptor angiotensin-converting enzyme 2 (ACE2) in brain organoids are substantially lower than in the lung or intestinal epithelium. In the brain, alternative receptors, such as CD147 and DPP4, are required for infection and virus replication as shown in astrocytes88.
Similarly, single-cell RNA sequencing (scRNA-seq) of kidney organoids showed that SARS-CoV-2 directly infects kidney cells, triggering pro-fibrotic events and features of polycystic kidney disease, closely resembling renal processes observed in individuals who are critically ill with COVID-19 (refs. 89,90). Furthermore, transcriptomic profiling of infected kidney organoids aligned with the proteomic profile of the urine of individuals who were critically ill with COVID-19, further validating the findings of the model.
Similar examples can be found for other diseases such as hepatitis B91, allergic asthma92, chlamydia93 and influenza94,95. A true scientific leap was the establishment of an in vitro cultivation method for human norovirus in 2016 (ref. 96). Prior to that, numerous attempts to cultivate norovirus in intestinal epithelial cells or primary immune cells had failed owing to a lack of mimicry of the natural host environment, thereby preventing a full mechanistic understanding of norovirus infections96. Infection studies using human ASC-derived intestinal organoids provided the solution as they closely emulate the host environment by differentiating into physiologically active, multicellular epithelial tissues. Eventually, enterocytes were identified as the primary target for viral infection, which is pivotal when aiming, for example, for drug target identification. Ultimately, this discovery laid the foundation for numerous ongoing efforts to develop preventive or therapeutic97 measures, including a norovirus vaccine98.
Human-based models also contributed to an in-depth understanding of the mechanisms of infection of the Zika virus, which caused an outbreak in 2015 in South America. Rodent models cannot fully reproduce the mechanism by which the virus causes microcephaly because the human brain has an additional cortical layer containing radial glial cells (progenitor cells). However, infection studies in brain organoids showed that the Zika virus primarily infects neural progenitor cells, which increases neural cell death and reduces proliferation, thereby hampering neurogenesis and causing microcephaly99,100. This breakthrough provided the community with an experimental platform to screen for therapeutic options.
A recent discovery of similar magnitude is that infecting intestinal organoids with strains of Escherichia coli that produce the genotoxin colibactin revealed mutation signatures that could drive colorectal cancer (CRC) progression, similar to those found in human tumours101. Specifically, genotoxic E. coli strains were found to cause single base substitutions and indel formations in the intestinal epithelium and other tissues such as the urinary tract. These findings could have broad implications; for example, the detection and eradication of genotoxic E. coli strains could decrease the risk of intestinal cancer in large cohorts. Furthermore, the E. coli strain Nissle 1917, commonly used as a probiotic, also produces colibactin102, warranting re-evaluation of its use.
OoC devices can be used to study the pathomechanism of diseases that involve two or more organs. For example, the gut–liver–brain axis has long been postulated to contribute to Parkinson disease through short-chain fatty acids (metabolites of the intestinal microbiome) that can, directly and indirectly, promote neurodegeneration. This connection has been confirmed using an OoC setup that emulates the immune–metabolic crosstalk between these tissues103. Interestingly, co-cultivation of liver, gut, and a mixture of neurons, astrocytes and microglia facilitated the maturation of the brain model, which is difficult to achieve in single-tissue models. To emulate the disease state, iPS cells derived from individuals with Parkinson disease were differentiated into a cerebral model, co-cultured with a healthy gut and liver model, and exposed to circulating regulatory T and T helper 17 cells. Multi-omics and multiplexed cytokine and chemokine analysis revealed that short-chain fatty acids increase the expression of pathways related to ferroptosis in Parkinson disease models, a well-established cause of dopaminergic cell death in Parkinson disease. Animal models are not amenable to decoupling the effect of single parameters with such accuracy. Another example is an OoC model of the neurovascular unit infected by Cryptococcus neoformans, a fungus that causes fungal meningitis. This system revealed that C. neoformans trespasses the blood–brain barrier through transcytosis, providing a potential therapeutic target to inhibit this process104.
Drug screening and efficacy testing in 2D
Advances in patient-derived iPS cells have re-established the role of 2D cultures in preclinical drug development, leading to several breakthroughs in neurodegenerative diseases, conditions that have been historically plagued by the highest failure rates (≥97%) in drug development105. Since 2011, five approved first-in-class drugs for neurodegenerative diseases were identified through phenotypic screening of 2D cultures105. In phenotypic screens, the readout is based on alterations of the phenotype of a diseased cell or tissue (model), whereas no specific drug target is known. These screens, which are typically conducted in genetically engineered cell lines that harbour the target of interest, led to the discovery of the splice modulators risdiplam106 and branaplam107 for the treatment of spinal muscular atrophy, an autosomal recessive disease characterized by the degeneration of motor neurons. The efficacy of the lead compounds was assessed in iPS cell-derived motor neurons showing splice correction and restored protein levels. Risdiplam received its marketing authorization in the EU in 2021 and became the first orally available medication for spinal muscular atrophy.
Combinations of phenotypic screens with multi-omics technologies can further increase the success in early phases of drug development, especially for diseases with a complex genetic background10. For example, a screen of small molecules that correct dysregulated gene networks in NOTCH1-deficient calcific aortic valve disease (CAVD) was combined with a machine learning algorithm that was trained to classify gene expression levels as wild type or diseased, allowing the detection of target molecules in patient-derived iPS cells108. Almost 1,600 molecules were screened using targeted RNA sequencing, resulting in the identification of an inverse agonist of oestrogen-related receptor-α as the lead compound, which proved effective in correcting dysregulated CAVD-relevant genes in primary aortic valve endothelial cells from individuals with CAVD and in genetically modified mice.
Drug screening and efficacy testing in 3D
During the COVID-19 pandemic, 3D human-based infection models proved extremely valuable for the identification of new antiviral strategies82,109 and drug repurposing with SARS-CoV-2-dampening effects51,110,111,112. For example, high-throughput screens of ≥1,000 drugs approved by the FDA run on colonic and lung organoids identified several SARS-CoV-2 entry inhibitors such as imatinib, mycophenolic acid and quinacrine dihydrochloride111. Similarly, among eight drugs with dose-dependent inhibition of virus uptake in static Huh-7 cell monolayers, only three (amodiaquine, toremifene and clomiphene) proved effective in 3D OoC cultures51. Building on these results, amodiaquine (for example, NCT04532931, NCT04502342)51 and imatinib111 (for example, NCT04394416) have entered clinical trials.
Interestingly, applying physiological breathing mechanics in alveoli-on-chip models showed antiviral activity by increasing the expression of interferon-related genes and improving host defence113. For example, an increase in S100A7 expression was observed which codes for alarmins and receptor for advanced glycation end products (RAGE) ligands. The RAGE inhibitor azeliragon was then identified as effective by suppressing the overshooting of inflammatory responses as observed in patients who were severely ill with COVID-19. This data has been submitted as part of a pre-investigational new drug application to the FDA. Notably, the antimalarial drugs chloroquine and hydroxychloroquine, which received controversial attention during the COVID-19 pandemic, proved non-effective in a human lung-on-chip disease model, similar to clinical findings51.
Major advances against cancer have also been achieved using human disease models. For example, MCLA-158, a bispecific antibody binding epidermal growth factor receptor (EGFR) and leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5), was identified as the most effective antibody against wild-type and KRAS-mutant CRC after screening a large biobank of patient-derived CRC organoids. Importantly, MCLA-158 reliably discriminated between cancerous and healthy cells. These findings relied entirely on human-derived cancer organoids and went from bench to bedside in just 3 years (NCT03526835)114.
Similarly, the efficacy of amivantamab, a bispecific antibody against EGFR and the mesenchymal–epithelial transition receptor, was preclinically tested in patient-derived cells and organoids that harbour EGFR Exon20ins mutations115. Patients with this mutation have a poor prognosis because standard drugs, such as tyrosine kinase inhibitors, are ineffective. These results were replicated in clinical trials (for example, NCT04538664), eventually leading to the approval of amivantamab by the EMA for the treatment of non-small cell lung cancer.
Another successful example is the use of OoC models of chronic inflammatory demyelinating polyneuropathy, an autoimmune disease that cause muscle weakness, conduction blocks and aberrant spinal reflexes116. An OoC-based chronic inflammatory demyelinating polyneuropathy electrical conduction model, consisting of human iPS cell-derived motor neurons and Schwann cells cultivated on microelectrode arrays, closely emulates clinically relevant features such as muscle contraction and electrical activity. Treatment with the antibody TNT005, a specific complement component 1s inhibitor, inhibited the immune reaction and rescued the functional deficits, prompting the FDA to approve clinical trials (NCT04658472).
Clinical research
Beyond preclinical research, human disease models are also revolutionizing clinical decision-making. A prime example is cystic fibrosis, a heterogeneous genetic disease with limited treatment options and highly variable treatment outcomes depending on the disease-causing mutation (≥2,000 mutations reported). Cystic fibrosis is caused by loss-of-function mutations in the CFTR gene resulting in highly viscous mucus, which blocks the airways, limits the ability to breathe and causes persistent lung infections. The life expectancy of a newborn with cystic fibrosis in high-income countries is 55 years today.
A breakthrough in the treatment of cystic fibrosis was the development of CFTR modulators such as ivacaftor or lumacaftor — effective but also expensive drugs. Ivacaftor is registered for the treatment of patients with nine CFTR gating mutations, which make up only 5% of all individuals with with the disease, although combinations with other modulators can improve clinical responses and patient eligibility117.
However, identifying potential responders is difficult, especially for patients with rare mutations. To overcome this limitation, testing of treatment responses in intestinal patient-derived organoids (PDOs) has emerged as a reliable and predictive approach118,119. For example, rectal organoids from 71 individuals with cystic fibrosis that harboured 28 different CFTR mutations revealed residual CFTR function and identified effective drug combinations. In this case, measuring the lumen area of the organoids allowed comparison of CFTR function among patients, thereby providing physicians with a reliable predicting tool for clinical decision-making.
Similar applications are being pursued with cancer PDOs120,121,122, revealing strong correlations between in vitro and in vivo drug responses against cancer in the gastrointestinal tract123, bladder124, ovary125, rectum126 and pancreas127,128. For example, a living biobank of cancer PDOs from patients with metastatic and pre-treated colorectal and gastroesophageal cancer shows strong morphological, genotypic and spatiotemporal similarities between the primary tumour and the PDOs123. Furthermore, PDOs reliably predicted treatment responses in patients, with 100% sensitivity, 93% specificity, 88% positive predictive value and 100% negative predictive value, including response to taxanes and anti-EGFR antibodies, drugs that lack biomarkers to predict responsive patient subsets.
Similar results were obtained for pancreatic ductal adenocarcinoma, another aggressive and difficult-to-treat tumour with high recurrence rates. Traditionally, treatment decisions are based on the performance status and comorbidities of the patient but there is an unmet medical need for patient stratification. To address this issue, 114 PDO cultures from 101 patients were generated127 and subjected to transcriptomic profiling, followed by therapeutic profiling through treatment with the five most commonly used chemotherapeutics, including gemcitabine, nab-paclitaxel, irinotecan, 5-fluorouracil and oxaliplatin. The treatment outcome of the PDOs positively correlated with individual patient responses and the assessment of targeted therapy sensitivity can guide patient-specific treatment decisions. These studies demonstrate that implementing PDOs in clinical decision-making can directly benefit patients with cancer.
Human disease models have also helped to identify treatment-enhancement strategies, for example, using cyclin-dependent kinase inhibitors to increase the effectiveness of immune-checkpoint inhibitors129. Despite being very effective, only a subset of patients respond to checkpoint blockade therapy and the underlying mechanisms for resistance development are poorly understood. To enhance the treatment efficacy, a combinatory approach with small-molecule kinase inhibitors has been proposed to facilitate immune reactivation. Interestingly, cyclin-dependent kinase inhibitors 4 and 6 (CDK4/6) facilitate T cell activation, resulting in a higher number of tumour-infiltrating T cells in PDOs, cultivated over several days in a microfluidic 3D chip setup. CDK4/6 inhibition also showed high in vitro and in vivo activity and synergy with anti-PD-1 blocking antibodies129. Several clinical trials (such as NCT04799249, NCT03294694 and NCT04213404) are currently assessing the clinical translatability of these findings.
Overall, ClinicalTrials.gov currently lists 131 studies for the search term ‘organoids’, most of which focus on cancer. Most of these studies have generated PDOs and compared drug effects in vitro and in vivo. Out of these, 10 studies are already using PDOs for clinical decision-making (Table 1). No results were retrieved for the search term ‘organ-on-chip’ or ‘microphysiological system’.
Future applications in clinical trials
Patient-specific disease models hold great potential for personalized and precision medicine in which therapeutic decisions or interventions are tailored to the individual based on their genetic makeup, disease or potential side effects. Such patient-stratified approaches could also facilitate clinical drug testing, for example, in phase I clinical trials, when the pharmacokinetic and drug safety profile are usually assessed in ≤100 healthy individuals. Women are often underrepresented, partly owing to the risk of reproductive toxicity. This is highly problematic as there are sex-related differences in how men and women absorb, distribute and metabolize drugs, ultimately affecting drug efficacy and safety48,130,131. Human-based models from both sexes and different ethnicities could help pinpoint these inter-individual differences, thereby improving therapeutic assessment. Moreover, human (disease) models can be leveraged to pre-screen patient subpopulations to identify those that could benefit most from a new drug.
Framework for disease model design
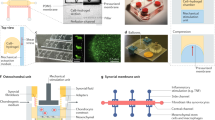
Defining universally applicable guidelines can be helpful to generate a predictive disease model. Although frameworks have been proposed for organoid and OoC setups24,54, providing a universal disease model design is challenging because their usefulness is strongly context dependent and influenced by the intended use and relevant readout parameters (Fig. 3). Furthermore, logistic requirements, such as access to relevant expertise, cell sources, equipment and cost, also need to be considered.

The flow chart shows the most important design considerations and workflows required for bioengineering human disease model. ECM, extracellular matrix.
When designing a disease model, the choice between model type (for example, 2D versus 3D or organoid versus bioengineered model), the selection of a suitable experimental setup (static versus dynamic) and the identification of important cell types are of utmost importance for the model to function. For drug screening, for example, 2D high-throughput methods using patient-specific or genetically engineered cells are most suitable as they allow testing of a large number of compounds in a short time. If the goal is to unravel disease mechanisms or study drug efficacy and safety, disease models with higher biomimicry and complexity are required. For example, when studying systemic conditions such as inflammation or metastatic cancer, complex multi-OoC setups are superior to single-tissue or organ models. Building on previously discussed examples, three key design considerations can be extracted, which are summarized in this section.
Cell diversity and tissue biomimicry
The identification of disease-relevant and tissue-relevant cell types is crucial, a prime example being the identification of the Zika virus tropism towards neural progenitor cells in brain organoids99. Equally important are the cultivation conditions, showcased by the decade-long failure in cultivating the norovirus ex vivo. Initial cultivation attempts, for example, in transformed epithelial cell lines and primary immune cells, failed largely because important factors of the host intestinal milieu were missing96. Furthermore, relevant biological barriers, such as the blood–brain barrier or the mucus hydrogel of the bronchial or intestinal epithelium, need to be included in the model. These are the primary defence mechanism of the human body and thereby very restrictive, especially in terms of the cut-off size of molecules that can pass. Failing to integrate them would ultimately limit the predictive value of disease models.
Model scaling
Scaling refers to maintaining proper ratios in size and rate parameters among different modules of a disease model, allowing the translation of in vitro data to clinical applications in vivo. Its importance has been recognized early on, but a general principle is still lacking132,133,134. Proper scaling needs to account for the geometry of the model, spatial arrangement of cells and operating conditions, the latter including medium dosage and perfusion rates of nutrients and metabolites, among other factors. There are two main principles of scaling: direct scaling, which uses the size ratio between a human body and the disease model to scale the size of each organ down to its in vitro counterpart55,135, and allometric scaling, which instead assumes that the size of organs and key physiological indices, such as heart, blood flow and metabolic rates, scale with the body mass according to power laws, with different exponents for different organs136,137. These two methods account for length and mass scales but not the time scale, and are often inaccurate. For example, the medium circulation rate is disproportionately slow compared to physiological blood flow, which in turn slows down the transport of nutrients and metabolic processes in tissue models138.
Therefore, dynamic, time-dependent operating conditions are more difficult to scale. Miniaturized models typically carry fewer cells and lack a pervasive vasculature to perfuse the tissue compared to actual organs. Thus, the perfusion, diffusion, permeation, nutrient and metabolic rates as well as their dosage need to be carefully calculated to ensure physiological relevance138. For this purpose, functional scaling136,138,139 seeks to match a key functional index between the model and the target organ in vivo. Such an index can be, for example, the drug clearance rate for the liver or the filtration rate for the kidney. However, it has proven difficult to balance competing key indices in multi-OoC setups; for a gut–liver system139, for example, a perfusion rate chosen for the gut to yield proper metabolic rates might result in drug exposure times in the liver that are too short.
To overcome these challenges, the similarity scaling approach140 adapts engineering techniques of dimensional analysis and similitude to OoCs. This strategy accounts for different similarity criteria (Box 1) simultaneously in a systematic framework. In any system, if an output is determined by a list of input variables and parameters, the latter must be algebraically linked in such a way as to yield the correct dimension or unit for the output. This constrains the algebraic relationship between the inputs and the output. Mathematically, the constraint is represented by combining the output and inputs into dimensionless groups141. Scaling an in vitro disease model with an in vivo organ boils down to matching the dimensionless groups between the two (Fig. 3). For an OoC, this matching could consist of proper ratios of the geometric lengths, morphologically correct arrangement of the cell types, and suitable ratios of mechanical and metabolic rates. These considerations ensure the correct translation of the key functional indices from the model to in vivo conditions139.
Biomechanical cues
Biomechanical stimuli, such as shear stress, stretching or compression, influence (patho)physiological processes142,143,144,145,146,147. A sequential and physiological exposure to biomechanical cues promotes the differentiation and maturation of organoids and bioengineered tissue models. For example, intestinal stem cells respond to the stiffness of the surrounding matrix, which guides cell differentiation and organoid formation148. Similarly, exposure to mechanical forces that mimic cardiac preload and afterload improves the contractility, cell alignment and conduction velocity of bioengineered heart tissue149. Another example using an alveoli-on-chip model revealed in vivo-like pathological responses to IL-2 toxicity146 or nanoparticle inhalation143 only when breathing mechanics were applied. Similarly, engineered heart tissues generated from patient-derived cells that harbour a desmoplakin mutation develop clinical arrhythmogenic cardiomyopathy only when exposed to dynamic mechanical loading149. In vivo, mutations in the desmoplakin gene cause a specific type of cardiomyopathy, which is characterized by a thickening and stiffening of the heart muscle resulting in cardiac dysfunction. These examples highlight the importance of applying disease-relevant and tissue-relevant biomechanical cues in bioengineered models. This consideration directly feeds into the choice of model type as not all of them allow the integration of physical forces per se.
Challenges and limitations
Despite fast-paced advances, several challenges remain that limit the widespread application of bioengineered disease models.
Limited complexity
Animal models are the only preclinical systems that allow the study of disease mechanisms and drug effects in the complex environment of a living organism. This argument is also commonly used to justify their necessity in biomedical research. Although this statement is not wrong per se, it fails to answer questions such as how valuable it is to know that a drug candidate is safe and effective in an inbred mouse strain (given the high rate of translational failure) or what it means for humans that certain nanoparticles can target the kidneys of zebrafish. Although some animal models mimic human physiology and/or pathology better than others (for example, the lungs and skin of pigs are anatomically and physiologically similar to human lungs and skin, which is not the case for rodents), simply conducting experiments in a living organism might not hold the solution.
Nonetheless, similar unresolved questions can be asked when developing bioengineered human models, namely, how complex is complex enough? Or how simple can a model be and still remain predictive of human pathophysiology? Putative key components, such as cells or biomechanical cues relevant to the disease of interest, are included based on existing knowledge, yet the possibility to overlook and thus exclude disease-relevant contributors remains. This biased approach bears the risk of false-positive or false-negative results.
Limited lifespan and long cultivation
Although some OoC models have been maintained for up to 3 months55, the effective lifetime of most disease models spans over a few days. The limited regenerative capacity of primary cell-based models is a major limitation as is identifying a universal culture medium that can maintain tissues of different germ layers in multi-organ models. Media mixtures are typically used in these models, which, however, could reverse tissue maturation because cells from different germ layers are exposed to unspecific factors. This mismatch could result in gene and protein expression levels that are atypical for the tissue of interest, a feature that seems particularly relevant for stem cell-derived models. Generating tissue-specific niches by introducing native endothelial barriers in OoC setups might hold a solution for this problem; while these barriers keep the tissue models in their optimized environment by separating them from a common media circuit, inter-tissue communication is still possible through the exchange of, for example, cytokines or exosomes150.
Notably, organoids can be cultivated over months and probably even years, thus enabling the investigation of tissue maturation in a disease context as shown for SARS-CoV-2 infections in brain organoids87. However, even such a long lifespan might not be sufficient to study neural diseases that develop or progress slowly — for example, Alzheimer disease and Parkinson disease. In this case, the simplicity of bioengineered human disease models could prove advantageous because disease-relevant effects might appear more rapidly owing to the lack of compensatory mechanisms that typically exist in vivo that can compensate for or mask tissue dysfunction.
Moreover, broad clinical implementation of patient-specific disease models, such as PDOs, is currently hindered by low efficiencies in PDO establishment (60–70% success rate on average although, for some tissues, up to 90% have been reported for intestinal organoids151) as well as lengthy generation times and expansion procedures. The establishment and expansion of PDOs and subsequent drug testing currently span several weeks or even months, although technical solutions to shorten the process are emerging. For example, microfabricated array devices using organoids at passage 0 provide drug screening results within a week without compromising the predictivity of patient responses to anticancer drugs152.
Similarly, protocols for cell differentiation into disease models can span several weeks, especially for multicellular tissue models. To accelerate and guide stem cell differentiation, the overexpression of transcription factors relevant to the differentiation of the target tissue (for example, ETV2 for endothelial cells or NGN1 for neurons) can switch on rapid cell differentiation. Subsequent mixing or controlled spatial patterning through, for example, 3D printing of iPS cells pre-programmed for doxycycline-induced overexpression of such cell type-specific transcription factors, ultimately facilitates the differentiation into multicellular and spatially patterned organoids independently from extracellular differentiation cues present in the culture medium153. This strategy could substantially expedite the generation of multicellular disease models.
Immunoregulation
Emulating the fine-tuned and highly interdependent immunoregulation in the human body remains a main limitation of in vitro human disease models. The human immune system is incredibly complex, orchestrating a multitude of immune cell (sub-)types of different functionality, many poorly understood and others probably still unrecognized. Although many disease models already contain certain immune cell types143,145,154,155,156, most models rely on the integration of only a few, pre-defined immune cell types, which, however, does not emulate the complexity of in vivo immunoregulation. For example, in the gut–liver–brain OoC setup for modelling Parkinson disease, only regulatory and T helper 17 cells were used103. Similarly, to study pathogen–host interactions in human disease models, only T cells and in some cases B cells have often been included113. By contrast, peripheral blood mononuclear cells have been increasingly added in OoC setups157. Bioengineered secondary immune organs, such as lymphoid tissue models158,159,160, might help overcome this limitation; however, this is still an underdeveloped area.
Disease model validation
A key task for the bioengineering community is to demonstrate the equivalence or superiority of human disease models over their animal-based counterparts. Therefore, rigorous disease model validation is essential but often neglected. The problem is further aggravated by variabilities in model composition, complexity and experimental protocols. First, the model needs to capture relevant features of the disease through, for example, histological similarity, in vivo-like gene regulation and protein expression patterns, or known drug responses. Similarly, the assays and readout parameters need to be relevant to the disease of interest, and the model and experimental setup should minimize noise and bias. Finally, it needs to be determined if the data generated by the model can be extrapolated for clinical applications. Establishing these criteria and ensuring inter-laboratory reproducibility needs to be standardized. Automated procedures ranging from liquid handling and cell seeding to sampling and sample analysis can improve the reproducibility, controllability and, thus, robustness of the models.
Omics technologies
Single-cell and spatial profiling at transcriptional, proteomic and epigenetic levels (Box 2) can substantially improve disease model validation161,162. For example, scRNA-seq helped identify and mitigate shortcomings related to the high variability of cell composition of in vitro models, exemplified in cerebral organoids generated from individuals with autism spectrum disorder163. By using an integrated scRNA and bulk RNA-seq approach, genes that showed low inter-individual variability, and thus high inter-individual correlation, allowed pinpointing of genes of relevance for autism spectrum disorder. Furthermore, combining scRNA-seq with ATAC-seq164 (Box 2) can help determine the degree of biomimicry of (disease) models by identifying cell type-specific chromatin accessibility patterns as well as transcription factors and cascades that are pivotal for cell fate specification and topographic identity as shown for brain and retinal organoids165,166. Using omics technologies, differences in gene regulation and signalling pathways between organoids and the corresponding primary tissue samples can be identified.
Imaging and data-driven approaches
The complex architecture of disease models, their characterization and the investigation of cell-specific pathological reactions require advanced 3D imaging technologies, such as light-sheet fluorescence microscopy or multiphoton imaging167. Unlike 2D imaging, these technologies allow high-resolution 3D whole-mount imaging of disease models at different scales, including cellular compositions, cell shapes, cell–cell interactions and cell fate167. The superiority of light-sheet fluorescence microscopy over conventional line-scan imaging has already been demonstrated for 3D organoids168. However, the huge amount of data generated, especially for high-throughput screenings of PDOs, has called for an increasing use of artificial intelligence-driven data analysis algorithms such as machine learning169. For example, tracking morphological and textural changes of PDOs to different drugs using bright field images enables the generation of PDO-specific dose–response curves170. Similarly, using a neural network-based high-throughput approach for light microscopy-based screening demonstrates strong correlations between clinical and predicted PDO response across different tumour entities171. Similarly, integration of network modelling and perturbation analysis with pathological disease features of 1,300 patient-derived brain organoids provided a high-content system for drug screening172. These combinatorial approaches could help identify effective drugs more reliably, although their actual impact has yet to be proven.
When aiming for routine and standardized applications, following the ‘digital twins’ concept can be useful. A digital twin is an in silico method that uses real-world data to predict how a product or process will perform. Examples of their successful application can be found in space missions or aeroplane construction173. In bioengineering, the implementation of digital twins could facilitate the shift from exploration towards a patient-focused and manufacturing-focused, and therefore more standardized, approach. For example, digital twin-based mathematical models should accurately describe real-world phenomena, starting with a simple structure and known effects (such as cell kinetics, stationary and fluid flow characteristics, and distribution of components such as growth factors) and gradually adapting it to newly available data and observations (for example, by including mass transfer and shear effects). Although still in their infancy, digital twins are being explored for tissue models and have enormous potential to be used for other disease models174,175.
Commercialization and accessibility
Developing and maintaining bioengineered disease models require high-level expertise and costly infrastructures. Even static 3D tissue models are substantially more expensive and technically demanding than conventional 2D biomedical methods. Moreover, the notion that in vitro methods are cheaper is misleading because bioengineered disease models can be as if not more expensive than animal models. As the demand for human-based models increases, more companies are emerging to fill the gap; however, commercially available setups are often still very expensive and therefore not affordable for many laboratories. Moreover, these models mainly focus on preclinical safety testing and not disease modelling and are not customizable169. For example, all commercially available OoC setups are run with pre-set chips, which determines a priori the type and dimension of tissues that can be integrated. Some OoC setups have the size of a coin (or even smaller), thereby making it difficult to cultivate actual 3D tissue models. Moreover, as of now, only few disease models are commercially available.
Outlook
Bioengineered disease models are revolutionizing biomedical research and will increasingly replace animal models in basic and preclinical research. Their implementation in preclinical or clinical stages could accelerate the drug development process, reduce false-positive and false-negative results, and facilitate the clinical translation of findings from bench to bedside. These benefits could also substantially cut down drug development costs, with an estimated 10–26% cost reduction per newly approved drug2. It is therefore not surprising that pharmaceutical companies have considerably ramped up their investments in this area.
Nonetheless, animal models are still considered the gold standard and therefore are often requested by default by grant review panels or reviewers. In our view, this is benchmarking against the wrong standard. Ideally, patient-derived in vivo data should be used as a benchmark to corroborate findings from human disease models, especially during model development and validation. Therefore, models should be continuously re-assessed to ensure that they comply with the most recent and relevant scientific findings.
Animal testing is not going to be completely replaced in the near future for several reasons, one of them being that toxicity testing in animals is legally mandated before entering clinical trials. Therefore, the most realistic short-term and mid-term scenario is the complementary use of both human-based and animal models. Nonetheless, drugs can already enter clinical trials without providing animal-derived data if the in vitro data is compelling, no suitable animal models are available and a considerable medical benefit is expected. Moreover, the FDA Modernization Act 2.0 has further broadened the scope of accepted preclinical models and encourages scientists to test drug efficacy and safety in human models whenever possible. This landmark decision is expected to cause ripple effects worldwide.
Notably, different disease models are suitable for different applications; organoids, for example, are useful for high-throughput drug screenings, whereas OoCs are more applicable for drug safety and efficacy testing. On the one hand, personalized disease models are well suited for clinical decision-making and in clinical trials to diversify the patient cohorts with respect to ethnicity, sex or age. On the other hand, they are poorly suited for high-throughput screenings. To ensure a widespread preclinical and clinical application of human disease models, meeting the following key milestones is essential: definition of robust criteria for disease model validation, benchmarking against patient-derived in vivo data, regulatory approval and legal basis for the implementation of human disease models in the drug development process, and the establishment of scalable, robust and standardized manufacturing processes (Fig. 3).
For this purpose, the experience from the in vitro pro-arrhythmia assay (CiPA) initiative, launched in 2013 to improve the assessment of the pro-arrhythmic potential of new drugs, can prove useful. Members of the CiPA initiative include regulatory authorities such as the FDA or EMA, pharmaceutical companies, and academics. The initiative has defined sub-working groups that collaborate to reach pre-defined milestones and could serve as a role model for the widespread implementation of bioengineered models in (pre-)clinical research.
The formation of dedicated core facilities would also make human disease models more accessible and promote their usage. In this regard, a survey amongst scientists that do not use OoC setups revealed that the lack of ready-to-use-systems and production facilities as well as high entry barriers and costs are the main reasons for not employing OoCs169. Therefore, facilities that support researchers with hands-on training, ready-to-use tissue models or scientific advice would promote the widespread usage of human disease models, the Hubrecht Organoid Technology organization in Utrecht, The Netherlands, being one example. Similarly, ‘living biobanks’ can provide access to patient-derived cells and organoids from large patient cohorts, and have already been established for different cancers124,151,176,177,178, genetic diseases such as cystic fibrosis, infectious diseases, chronic obstructive pulmonary disease and inflammatory bowel diseases179.
It is unrealistic to assume that the complexity of a human can be fully recapitulated in vitro. At the same time, the ideal should be to reach maximal clinical mimicry. Although several challenges remain unsolved, initial drawbacks, such as the lack of cell diversity, vascularization and the ability to study tissue crosstalk, have already been largely overcome, highlighting the potential of human disease models to fundamentally change the future of biomedical research.
Change history
13 June 2023
A Correction to this paper has been published: https://doi.org/10.1038/s44222-023-00088-8
References
Adhikary, P. P., Ul Ain, Q., Hocke, A. C. & Hedtrich, S. COVID-19 highlights the model dilemma in biomedical research. Nat. Rev. Mater. 6, 374–376 (2021).
Franzen, N. et al. Impact of organ-on-a-chip technology on pharmaceutical R&D costs. Drug Discov. Today 24, 1720–1724 (2019).
IQVIA Institute. Global Trends in R&D 2022 (IQVIA Institute, 2021).
Golding, H., Khurana, S. & Zaitseva, M. What is the predictive value of animal models for vaccine efficacy in humans? The importance of bridging studies and species-independent correlates of protection. Cold Spring Harb. Perspect. Biol. 10, a028902 (2018).
Franco, R. & Cedazo-Minguez, A. Successful therapies for Alzheimer’s disease: why so many in animal models and none in humans? Front. Pharmacol. 5, 146 (2014).
Ingber, D. E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 23, 467–491 (2022).
Brennan, F. R. et al. Safety testing of monoclonal antibodies in non-human primates: case studies highlighting their impact on human risk assessment. mAbs 10, 1–17 (2018).
Rothgangl, T. et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 39, 949–957 (2021).
Muñoz-Fontela, C. et al. Animal models for COVID-19. Nature 586, 509–515 (2020).
Brooks, I. R. et al. Functional genomics and the future of iPSCs in disease modeling. Stem Cell Rep. 17, 1033–1047 (2022).
Jensen, C. & Teng, Y. Is it time to start transitioning from 2D to 3D cell culture? Front. Mol. Biosci. 7, 33 (2020).
Htwe, S. S. et al. Investigating NF-κB signaling in lung fibroblasts in 2D and 3D culture systems. Respir. Res. 16, 144 (2015).
DesRochers, T. M., Suter, L., Roth, A. & Kaplan, D. L. Bioengineered 3D human kidney tissue, a platform for the determination of nephrotoxicity. PLoS One 8, e59219 (2013).
Vepari, C. & Kaplan, D. L. Silk as a biomaterial. Prog. Polym. Sci. 32, 991–1007 (2007).
Pati, F. & Cho, D. W. Bioprinting of 3D tissue models using decellularized extracellular matrix bioink. Methods Mol. Biol. 1612, 381–390 (2017).
Stoltz, J. F., Zhang, L., Ye, J. S. & De Isla, N. Organ reconstruction: dream or reality for the future. Biomed. Mater. Eng. 28, S121–S127 (2017).
Hassanpour, A., Talaei-Khozani, T., Kargar-Abarghouei, E., Razban, V. & Vojdani, Z. Decellularized human ovarian scaffold based on a sodium lauryl ester sulfate (SLES)-treated protocol, as a natural three-dimensional scaffold for construction of bioengineered ovaries. Stem Cell Res. Ther. 9, 252 (2018).
Debels, H., Hamdi, M., Abberton, K. & Morrison, W. Dermal matrices and bioengineered skin substitutes: a critical review of current options. Plast. Reconstr. Surg. Global Open 3, e284 (2015).
Caddeo, S., Boffito, M. & Sartori, S. Tissue engineering approaches in the design of healthy and pathological in vitro tissue models. Front. Bioeng. Biotechnol. 5, 40 (2017).
Upadhyay, S. & Palmberg, L. Air-liquid interface: relevant in vitro models for investigating air pollutant-induced pulmonary toxicity. Toxicol. Sci. 164, 21–30 (2018).
Moniz, T., Costa Lima, S. A. & Reis, S. Human skin models: from healthy to disease-mimetic systems; characteristics and applications. Br. J. Pharmacol. 177, 4314–4329 (2020).
Khetani, S. R. & Bhatia, S. N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 26, 120–126 (2008).
Cantley, W. et al. Functional and sustainable 3D human neural network models from pluripotent stem cells. ACS Biomater. Sci. Eng. 4, 4278–4288 (2018).
Hofer, M. & Lutolf, M. P. Engineering organoids. Nat. Rev. Mater. 6, 402–420 (2021).
Du, Y. et al. Development of a miniaturized 3D organoid culture platform for ultra-high-throughput screening. J. Mol. Cell Biol. 12, 630–643 (2020).
Vonk, A. M. et al. Protocol for application, standardization and validation of the forskolin-induced swelling assay in cystic fibrosis human colon organoids. STAR Protoc. 1, 100019 (2020).
Spence, J. R. et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109 (2011).
McCracken, K. W. et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404 (2014).
Takebe, T. et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499, 481–484 (2013).
Huang, L. et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell–and patient-derived tumor organoids. Nat. Med. 21, 1364–1371 (2015).
Wong, A. P. et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat. Biotechnol. 30, 876–882 (2012).
Kurmann, A. A. et al. Regeneration of thyroid function by transplantation of differentiated pluripotent stem cells. Cell Stem Cell 17, 527–542 (2015).
Taguchi, A. et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14, 53–67 (2014).
Phillips, M. J. et al. Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Invest. Ophthalmol. Vis. Sci. 53, 2007–2019 (2012).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Mansour, A. A. et al. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 432–441 (2018).
Revah, O. et al. Maturation and circuit integration of transplanted human cortical organoids. Nature 610, 319–326 (2022).
Sachs, N. et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 38, e100300 (2019).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Boj, S. F. et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338 (2015).
Bartfeld, S. et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148, 126–136.e6 (2015).
Huch, M. et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015).
Turco, M. Y. et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 19, 568–577 (2017).
Drost, J. et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 11, 347–358 (2016).
Kessler, M. et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 6, 8989 (2015).
Linnemann, J. R. et al. Quantification of regenerative potential in primary human mammary epithelial cells. Development 142, 3239–3251 (2015).
Chichagova, V. et al. Human iPSC differentiation to retinal organoids in response to IGF1 and BMP4 activation is line- and method-dependent. Stem Cell 38, 195–201 (2020).
Lock, R. et al. A framework for developing sex-specific engineered heart models. Nat. Rev. Mater. 7, 295–313 (2021).
Lai Benjamin, F. L. et al. Recapitulating pancreatic tumor microenvironment through synergistic use of patient organoids and organ-on-a-chip vasculature. Adv. Funct. Mater. 30, 2000545 (2020).
Yadid, M. et al. Endothelial extracellular vesicles contain protective proteins and rescue ischemia-reperfusion injury in a human heart-on-chip. Sci. Transl. Med. 12, eaax8005 (2020).
Si, L. et al. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 5, 815–829 (2021).
Low, L. A., Mummery, C., Berridge, B. R., Austin, C. P. & Tagle, D. A. Organs-on-chips: into the next decade. Nat. Rev. Drug Discov. 20, 345–361 (2021).
Vunjak-Novakovic, G., Ronaldson-Bouchard, K. & Radisic, M. Organs-on-a-chip models for biological research. Cell 184, 4597–4611 (2021).
Leung, C. M. et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Primers 2, 33 (2022).
Maschmeyer, I. et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 15, 2688–2699 (2015).
Lai, B. F. L. et al. A well plate-based multiplexed platform for incorporation of organoids into an organ-on-a-chip system with a perfusable vasculature. Nat. Protoc. 16, 2158–2189 (2021).
Chou, D. B. et al. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat. Biomed. Eng. 4, 394–406 (2020).
Edington, C. D. et al. Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci. Rep. 8, 4530 (2018).
Herland, A. et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat. Biomed. Eng. 4, 421–436 (2020).
Novak, R. et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng. 4, 407–420 (2020).
Marx, U. et al. Biology-inspired microphysiological system approaches to solve the prediction dilemma of substance testing. Altex 33, 272–321 (2016).
Matsui, T. & Shinozawa, T. Human organoids for predictive toxicology research and drug development. Front. Genet. 12, 767621 (2021).
Li, M. et al. Advanced human developmental toxicity and teratogenicity assessment using human organoid models. Ecotoxicol. Environ. Saf. 235, 113429 (2022).
Cong, Y. et al. Drug toxicity evaluation based on organ-on-a-chip technology: a review. Micromachines 11, 381 (2020).
Nawroth, J. C. et al. Modeling alcohol-associated liver disease in a human liver-chip. Cell Rep. 36, 109393 (2021).
Sphabmixay, P. et al. High resolution stereolithography fabrication of perfusable scaffolds to enable long-term meso-scale hepatic culture for disease modeling. Biofabrication 13, ac23aa (2021).
Bauer, S. et al. Functional coupling of human pancreatic islets and liver spheroids on-a-chip: towards a novel human ex vivo type 2 diabetes model. Sci. Rep. 7, 14620 (2017).
Wimmer, R. A. et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565, 505–510 (2019).
Jabali, A. et al. Human cerebral organoids reveal progenitor pathology in EML1-linked cortical malformation. EMBO Rep 23, e54027 (2022).
Vatine, G. D. et al. Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell 24, 995–1005.e6 (2019).
Hönzke, S. et al. Influence of Th2 cytokines on the cornified envelope, tight junction proteins, and ß-defensins in filaggrin-deficient skin equivalents. J. Investig. Dermatol. 136, 631–639 (2016).
Zamprogno, P. et al. Second-generation lung-on-a-chip with an array of stretchable alveoli made with a biological membrane. Commun. Biol. 4, 168 (2021).
van der Vaart, J. et al. Modelling of primary ciliary dyskinesia using patient-derived airway organoids. EMBO Rep. 22, e52058 (2021).
Charbaji, R. et al. Design and testing of efficient mucus-penetrating nanogels — pitfalls of preclinical testing and leassons learned. Small https://doi.org/10.1002/smll.202007963 (2021).
Kayisoglu, O. et al. Location-specific cell identity rather than exposure to GI microbiota defines many innate immune signalling cascades in the gut epithelium. Gut 70, 687–697 (2021).
Lõhmussaar, K. et al. Patient-derived organoids model cervical tissue dynamics and viral oncogenesis in cervical cancer. Cell Stem Cell 28, 1380–1396.e6 (2021).
Vila, O. F. et al. Bioengineered optogenetic model of human neuromuscular junction. Biomaterials 276, 121033 (2021).
Williams, K. et al. A 3-D human model of complex cardiac arrhythmias. Acta Biomater. 132, 149–161 (2021).
Mastikhina, O. et al. Human cardiac fibrosis-on-a-chip model recapitulates disease hallmarks and can serve as a platform for drug testing. Biomaterials 233, 119741 (2020).
Wang, E. Y. et al. An organ-on-a-chip model for pre-clinical drug evaluation in progressive non-genetic cardiomyopathy. J. Mol. Cell Cardiol. 160, 97–110 (2021).
Ronaldson-Bouchard, K. et al. Engineering of human cardiac muscle electromechanically matured to an adult-like phenotype. Nat. Protoc. 14, 2781–2817 (2019).
Lu, R. X. Z. et al. Vasculature-on-a-chip platform with innate immunity enables identification of angiopoietin-1 derived peptide as a therapeutic for SARS-CoV-2 induced inflammation. Lab Chip 22, 1171–1186 (2022).
Zhang, M. et al. Biomimetic human disease model of SARS-CoV-2 induced lung injury and immune responses on organ chip system. Adv. Sci. 8, 2002928 (2020).
Katsura, H. et al. Human lung stem cell-based alveolospheres provide insights into SARS-CoV-2-mediated interferon responses and pneumocyte dysfunction. Cell Stem Cell 27, 890–904.e8 (2020).
Lamers, M. M. et al. An organoid-derived bronchioalveolar model for SARS-CoV-2 infection of human alveolar type II-like cells. EMBO J. 40, e105912 (2021).
Thacker, V. V. et al. Rapid endotheliitis and vascular damage characterize SARS-CoV-2 infection in a human lung-on-chip model. EMBO Rep. 22, e52744 (2021).
Ramani, A. et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 39, e106230 (2020).
Andrews, M. G. et al. Tropism of SARS-CoV-2 for human cortical astrocytes. Proc. Natl Acad. Sci. USA 119, e2122236119 (2022).
Jansen, J. et al. SARS-CoV-2 infects the human kidney and drives fibrosis in kidney organoids. Cell Stem Cell 29, 217–231.e8 (2022).
Helms, L. et al. Cross-validation of SARS-CoV-2 responses in kidney organoids and clinical populations. JCI Insight 6, e154882 (2021).
Ortega-Prieto, A. M. et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat. Commun. 9, 682 (2018).
Nawroth, J. C. et al. A microengineered airway lung chip models key features of viral-induced exacerbation of asthma. Am. J. Respir. Cell Mol. Biol. 63, 591–600 (2020).
Koster, S. et al. Modelling Chlamydia and HPV co-infection in patient-derived ectocervix organoids reveals distinct cellular reprogramming. Nat. Commun. 13, 1030 (2022).
Liberti, D. C. et al. Alveolar epithelial cell fate is maintained in a spatially restricted manner to promote lung regeneration after acute injury. Cell Rep. 35, 109092 (2021).
Bui, C. H. T. et al. Tropism of influenza B viruses in human respiratory tract explants and airway organoids. Eur. Respir. J. 54, 1900008 (2019).
Ettayebi, K. et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393 (2016).
Hayashi, T. et al. Dasabuvir inhibits human norovirus infection in human intestinal enteroids. mSphere 6, e0062321 (2021).
Alvarado, G. et al. Broadly cross-reactive human antibodies that inhibit genogroup I and II noroviruses. Nat. Commun. 12, 4320 (2021).
Qian, X. et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254 (2016).
Garcez, P. P. et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–818 (2016).
Pleguezuelos-Manzano, C. et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 580, 269–273 (2020).
Nougayrède, J. P. et al. A toxic friend: genotoxic and mutagenic activity of the probiotic strain Escherichia coli Nissle 1917. mSphere 6, e0062421 (2021).
Trapecar, M. et al. Human physiomimetic model integrating microphysiological systems of the gut, liver, and brain for studies of neurodegenerative diseases. Sci. Adv. 7, eabd1707 (2021).
Kim, J. et al. Fungal brain infection modelled in a human-neurovascular-unit-on-a-chip with a functional blood-brain barrier. Nat. Biomed. Eng. 5, 830–846 (2021).
Swalley, S. E. Expanding therapeutic opportunities for neurodegenerative diseases: a perspective on the important role of phenotypic screening. Bioorg Med. Chem. 28, 115239 (2020).
Naryshkin, N. A. et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693 (2014).
Palacino, J. et al. SMN2 splice modulators enhance U1–pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 11, 511–517 (2015).
Theodoris, C. V. et al. Network-based screen in iPSC-derived cells reveals therapeutic candidate for heart valve disease. Science 371, eabd0724 (2021).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913.e7 (2020).
Bai, H. et al. Mechanical control of innate immune responses against viral infection revealed in a human lung alveolus chip. Nat. Commun. 13, 1928 (2021).
Han, Y. et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 589, 270–275 (2021).
Rahmani, W. et al. Attenuation of SARS-CoV-2 infection by losartan in human kidney organoids. iScience 25, 103818 (2022).
Bai, H. et al. Mechanical control of innate immune responses against viral infection revealed in a human lung alveolus chip. Nat. Commun. 13, 1928 (2022).
Herpers, B. et al. Functional patient-derived organoid screenings identify MCLA-158 as a therapeutic EGFR × LGR5 bispecific antibody with efficacy in epithelial tumors. Nat. Cancer 3, 418–436 (2022).
Yun, J. et al. Antitumor activity of Amivantamab (JNJ-61186372), an EGFR-MET bispecific antibody, in diverse models of EGFR exon 20 insertion-driven NSCLC. Cancer Discov. 10, 1194–1209 (2020).
Rumsey, J. W. et al. Classical complement pathway inhibition in a “human-on-a-chip” model of autoimmune demyelinating neuropathies. Adv. Ther. 5, 2200030 (2022).
Fiedorczuk, K. & Chen, J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 378, 284–290 (2022).
Dekkers, J. F. et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci. Transl. Med. 8, 344ra384 (2016).
Berkers, G. et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 26, 1701–1708.e3 (2019).
Ooft, S. N. et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci. Transl. Med. 11, eaay2574 (2019).
Schütte, M. et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 8, 14267 (2017).
Shi, R. et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin. Cancer Res. 26, 1162–1174 (2020).
Vlachogiannis, G. et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926 (2018).
Lee, S. H. et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell 173, 515–528.e17 (2018).
Hill, S. J. et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 8, 1404–1421 (2018).
Yao, Y. et al. Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell 26, 17–26.e6 (2020).
Tiriac, H. et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 8, 1112–1129 (2018).
Driehuis, E. et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl Acad. Sci. USA 116, 26580–26590 (2019).
Deng, J. et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 8, 216–233 (2018).
Klein, S. L. & Flanagan, K. L. Sex differences in immune responses. Nat. Rev. Immunol. 16, 626–638 (2016).
Zeng, Z. et al. Sex-hormone-driven innate antibodies protect females and infants against EPEC infection. Nat. Immunol. 19, 1100–1111 (2018).
Ronaldson-Bouchard, K. & Vunjak-Novakovic, G. Organs-on-a-chip: a fast track for engineered human tissues in drug development. Cell Stem Cell 22, 310–324 (2018).
Park, D., Lee, J., Chung, J. J., Jung, Y. & Kim, S. H. Integrating organs-on-chips: multiplexing, scaling, vascularization, and innervation. Trends Biotechnol. 38, 99–112 (2020).
Malik, M., Yang, Y., Fathi, P., Mahler, G. J. & Esch, M. B. Critical considerations for the design of multi-organ microphysiological systems (MPS). Front. Cell Dev. Biol. 9, 721338 (2021).
Wagner, I. et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 13, 3538–3547 (2013).
Wikswo, J. P. et al. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip 13, 3496–3511 (2013).
Ahluwalia, A. Allometric scaling in-vitro. Sci. Rep. 7, 42113 (2017).
Sung, J. H., Wang, Y. & Shuler, M. L. Strategies for using mathematical modeling approaches to design and interpret multi-organ microphysiological systems (MPS). APL Bioeng. 3, 021501 (2019).
Maass, C., Stokes, C. L., Griffith, L. G. & Cirit, M. Multi-functional scaling methodology for translational pharmacokinetic and pharmacodynamic applications using integrated microphysiological systems (MPS). Integr. Biol. 9, 290–302 (2017).
Feng, J. J. & Hedtrich, S. A similarity scaling approach for organ-on-chip devices. Lab Chip 22, 3663–3667 (2022).
Zlokarnik, M. Scale-Up in Chemical Engineering 2nd edn (Wiley‐VCH Verlag GmbH & Co. KGaA, 2006).
Frye, M. et al. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nat. Commun. 9, 1511 (2018).
Huh, D. et al. Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668 (2010).
Sontheimer-Phelps, A. et al. Human colon-on-a-chip enables continuous in vitro analysis of colon mucus layer accumulation and physiology. Cell Mol. Gastroenterol. Hepatol. 9, 507–526 (2020).
Bein, A. et al. Enteric coronavirus infection and treatment modeled with an immunocompetent human intestine-on-a-chip. Front. Pharmacol. 12, 718484 (2021).
Huh, D. et al. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 4, 159ra147 (2012).
Grassart, A. et al. Bioengineered human organ-on-chip reveals intestinal microenvironment and mechanical forces impacting shigella infection. Cell Host Microbe 26, 435–444.e4 (2019).
Gjorevski, N. et al. Designer matrices for intestinal stem cell and organoid culture. Nature 539, 560–564 (2016).
Bliley, J. M. et al. Dynamic loading of human engineered heart tissue enhances contractile function and drives a desmosome-linked disease phenotype. Sci. Transl. Med. 13, eabd1817 (2021).
Ronaldson-Bouchard, K. et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 6, 351–371 (2022).
van de Wetering, M. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015).
Hu, Y. et al. Lung cancer organoids analyzed on microwell arrays predict drug responses of patients within a week. Nat. Commun. 12, 2581 (2021).
Skylar-Scott, M. A. et al. Orthogonally induced differentiation of stem cells for the programmatic patterning of vascularized organoids and bioprinted tissues. Nat. Biomed. Eng. 6, 449–462 (2022).
Wallmeyer, L. et al. TSLP is a direct trigger for T cell migration in filaggrin-deficient skin equivalents. Sci. Rep. 7, 774 (2017).
Maharjan, S., Cecen, B. & Zhang, Y. S. 3D immunocompetent organ-on-a-chip models. Small Methods 4, 2000235 (2020).
Kerns, S. J. et al. Human immunocompetent Organ-on-Chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies. eLife 10, e67106 (2021).
Cipriano, M. et al. Human immunocompetent choroid-on-chip: a novel tool for studying ocular effects of biological drugs. Commun. Biol. 5, 52 (2022).
Goyal, G. et al. Ectopic lymphoid follicle formation and human seasonal influenza vaccination responses recapitulated in an organ-on-a-chip. Adv. Sci. 9, e2103241 (2022).
Gibot, L. et al. Cell-based approach for 3D reconstruction of lymphatic capillaries in vitro reveals distinct functions of HGF and VEGF-C in lymphangiogenesis. Biomaterials 78, 129–139 (2016).
Gibot, L. et al. Tissue-engineered 3D human lymphatic microvascular network for in vitro studies of lymphangiogenesis. Nat. Protoc. 12, 1077–1088 (2017).
Li, X. & Wang, C.-Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral. Sci. 13, 36 (2021).
Kip, A. M. et al. Proteomics analysis of human intestinal organoids during hypoxia and reoxygenation as a model to study ischemia-reperfusion injury. Cell Death Dis. 12, 95 (2021).
Lim, E. T. et al. Orgo-Seq integrates single-cell and bulk transcriptomic data to identify cell type specific-driver genes associated with autism spectrum disorder. Nat. Commun. 13, 3243 (2022).
Baek, S. & Lee, I. Single-cell ATAC sequencing analysis: from data preprocessing to hypothesis generation. Comput. Struct. Biotechnol. J. 18, 1429–1439 (2020).
Finkbeiner, C. et al. Single-cell ATAC-seq of fetal human retina and stem-cell-derived retinal organoids shows changing chromatin landscapes during cell fate acquisition. Cell Rep. 38, 110294 (2022).
Ziffra, R. S. et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature 598, 205–213 (2021).
Fei, K., Zhang, J., Yuan, J. & Xiao, P. Present application and perspectives of organoid imaging technology. Bioengineering 9, 121 (2022).
Lallemant, L., Lebreton, C. & Garfa-Traoré, M. Comparison of different clearing and acquisition methods for 3D imaging of murine intestinal organoids. J. Biol. Methods 7, e141 (2020).
Busek, M., Aizenshtadt, A., Amirola-Martinez, M., Delon, L. & Krauss, S. Academic user view: organ-on-a-chip technology. Biosensors 12, 126 (2022).
Spiller, E. R. et al. Imaging-based machine learning analysis of patient-derived tumor organoid drug response. Front. Oncol. 11, 771173 (2021).
Larsen, B. M. et al. A pan-cancer organoid platform for precision medicine. Cell Rep. 36, 109429 (2021).
Park, J.-C. et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 12, 280 (2021).
Barricelli, B. R., Casiraghi, E. & Fogli, D. A survey on digital twin: definitions, characteristics, applications, and design implications. IEEE Access. 7, 167653–167671 (2019).
Möller, J. & Pörtner, R. Digital twins for tissue culture techniques — concepts, expectations, and state of the art. Processes 9, 447 (2021).
Fuhr, A., Kurtz, A., Hiepen, C. & Müller, S. Organoids as miniature twins — challenges for comparability and need for data standardization and access. Organoids 1, 28–36 (2022).
Yan, H. H. N. et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 23, 882–897.e11 (2018).
Sachs, N. et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 172, 373–386.e10 (2018).
Jacob, F. et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 180, 188–204.e22 (2020).
Ramalho, A. S. et al. Correction of CFTR function in intestinal organoids to guide treatment of cystic fibrosis. Eur. Respir. J. 57, 1902426 (2021).
Martin, L., Hutchens, M., Hawkins, C. & Radnov, A. How much do clinical trials cost? Nat. Rev. Drug Discov. 16, 381–382 (2017).
Werner, M. et al. Surface curvature differentially regulates stem cell migration and differentiation via altered attachment morphology and nuclear deformation. Adv. Sci. 4, 1600347 (2017).
Prodanov, L. et al. Long-term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol. Bioeng. 113, 241–246 (2016).
Peel, S. et al. Introducing an automated high content confocal imaging approach for organs-on-chips. Lab Chip 19, 410–421 (2019).
Abaci, H. E. & Shuler, M. L. Human-on-a-chip design strategies and principles for physiologically based pharmacokinetics/pharmacodynamics modeling. Integr. Biol. 7, 383–391 (2015).
Tsamandouras, N. et al. Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J. 19, 1499–1512 (2017).
Fleck, J. S. et al. Resolving organoid brain region identities by mapping single-cell genomic data to reference atlases. Cell Stem Cell 28, 1148–1159.e8 (2021).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Gonneaud, A. et al. A SILAC-based method for quantitative proteomic analysis of intestinal organoids. Sci. Rep. 6, 38195 (2016).
Cristobal, A. et al. Personalized proteome profiles of healthy and tumor human colon organoids reveal both individual diversity and basic features of colorectal cancer. Cell Rep. 18, 263–274 (2017).
Williams, K. E. et al. Quantitative proteomic analyses of mammary organoids reveals distinct signatures after exposure to environmental chemicals. Proc. Natl Acad. Sci. USA 113, E1343–E1351 (2016).
Melliou, S. et al. Regionally defined proteomic profiles of human cerebral tissue and organoids reveal conserved molecular modules of neurodevelopment. Cell Rep. 39, 110846 (2022).
Cong, Y. et al. Ultrasensitive single-cell proteomics workflow identifies >1000 protein groups per mammalian cell. Chem. Sci. 12, 1001–1006 (2021).
Williams, S. M. et al. Automated coupling of nanodroplet sample preparation with liquid chromatography–mass spectrometry for high-throughput single-cell proteomics. Anal. Chem. 92, 10588–10596 (2020).
Bakker, B. et al. Preparing ductal epithelial organoids for high-spatial-resolution molecular profiling using mass spectrometry imaging. Nat. Protoc. 17, 962–979 (2022).
Acknowledgements
We acknowledge partial funding of this work from the Stiftung Charité (S.H.), Einstein Foundation EC3R (A.L.), the Natural Sciences and Engineering Research Council Canada (NSERC, RGPIN-2020–04224 for S.H. and RGPIN-2019–04162 for J.J.F.), and the German Research Foundation (DFG)-funded Collaborative Research Centers 1449 (S.H., project A05) and 1340 (S.H., project A06).
Author information
Authors and Affiliations
Contributions
S.H., A.L. and J.J.F. contributed to the conceptualization, writing, figure drafting and revision of the article.
Corresponding author
Ethics declarations
Competing interests
The authors state no competing interests.
Peer review
Peer review information
Nature Reviews Bioengineering thanks Wei Zheng, who co-reviewed with Qi Zhang; Sina Bartfeld; Gordana Vunjak-Novakovic, who co-reviewed with Diogo Teles; and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Loewa, A., Feng, J.J. & Hedtrich, S. Human disease models in drug development. Nat Rev Bioeng 1, 545–559 (2023). https://doi.org/10.1038/s44222-023-00063-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44222-023-00063-3
This article is cited by
-
Ultrasound-assisted tissue engineering
Nature Reviews Bioengineering (2024)
-
Neuropathogenesis-on-chips for neurodegenerative diseases
Nature Communications (2024)
