Insights into the Kinetics of Intermediate Formation during Electrochemical Oxidation of the Organic Model Pollutant Salicylic Acid in Chloride Electrolyte

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
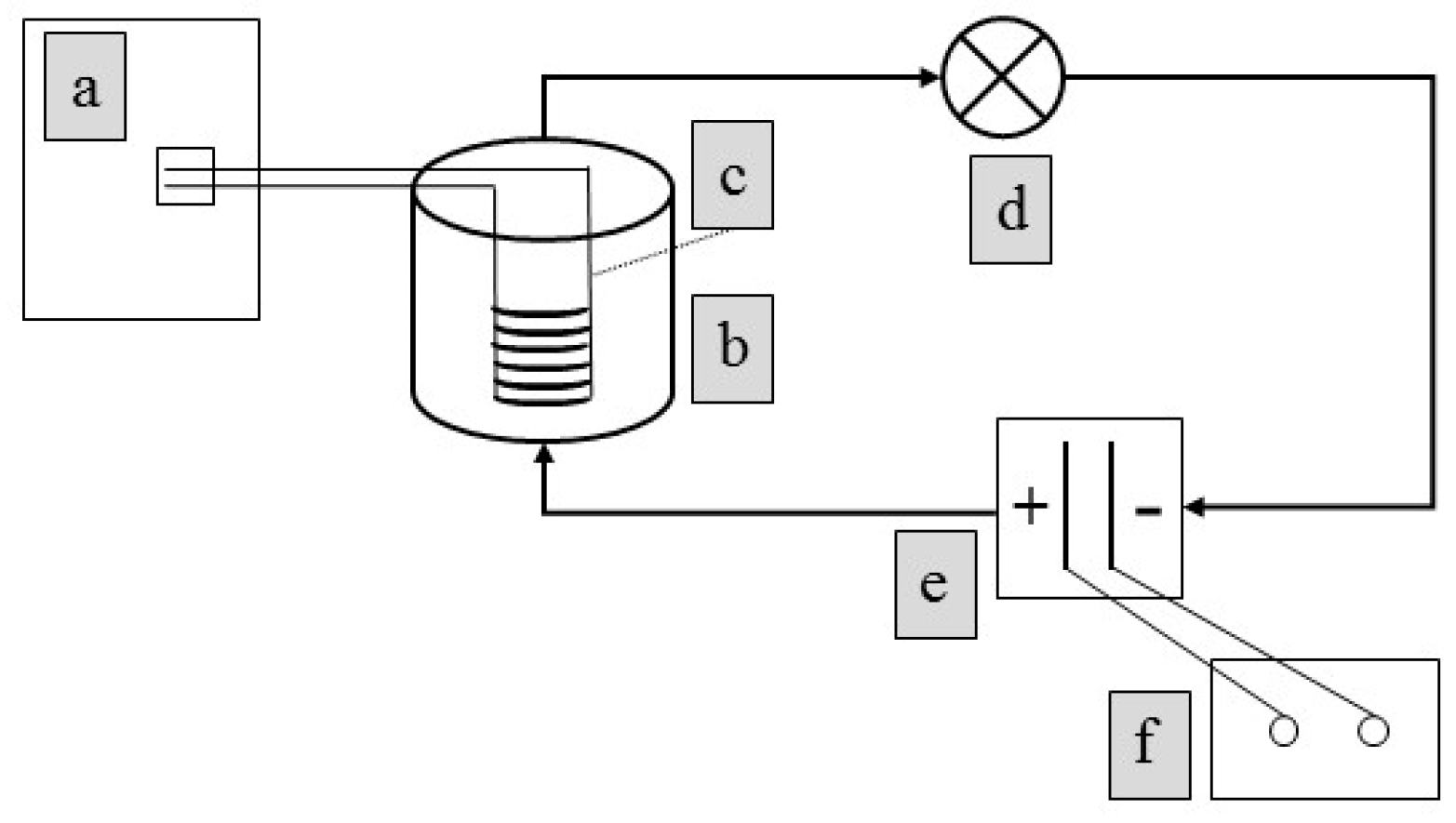
2.1. Cyclic Voltammetry
2.2. Bulk Electrolysis
2.3. Sample Analysis
2.4. Density Functional Theory Simulations
2.5. Kinetic Modelling
3. Results and Discussion
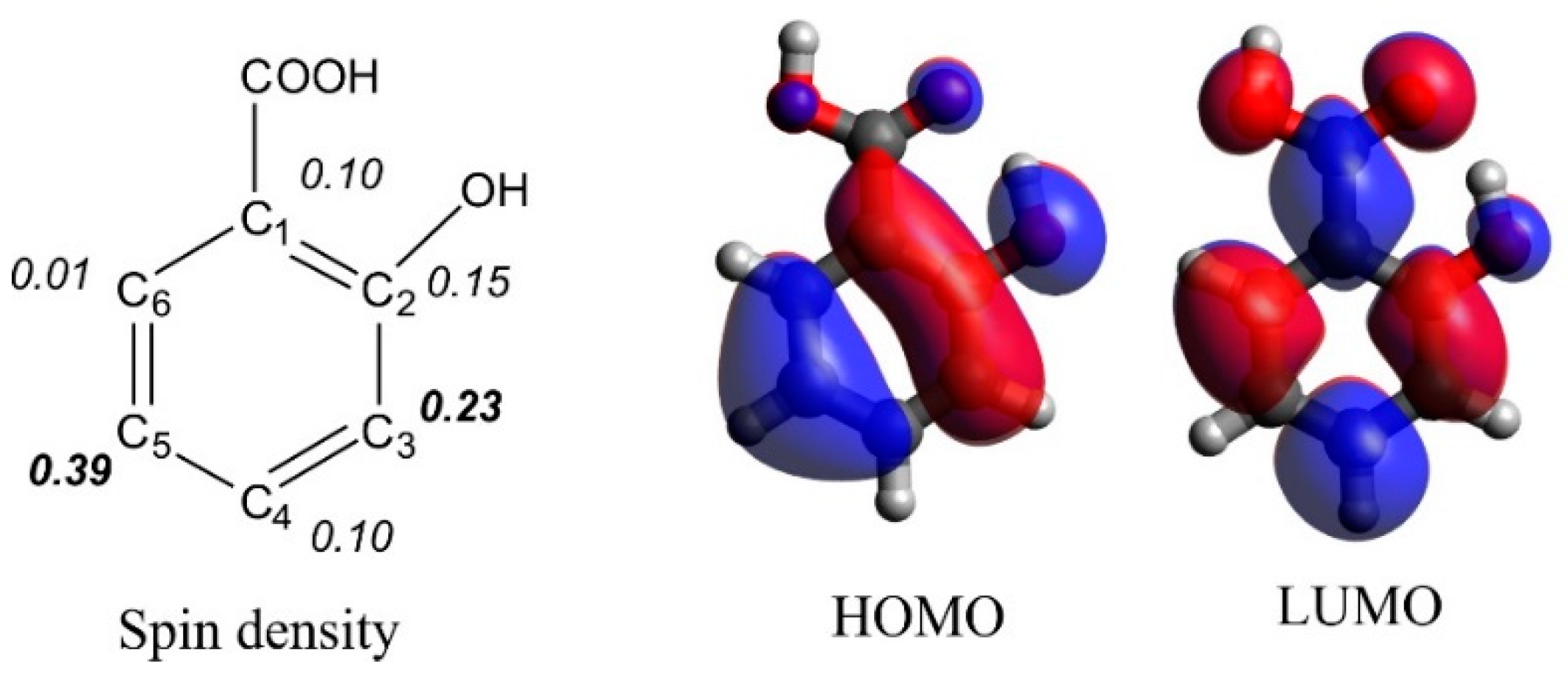
3.1. Prediction of Salicylic Acid Intermediate Formation by DFT Simulations
3.2. Oxidation of Salicylic Acid and Intermediate Formation
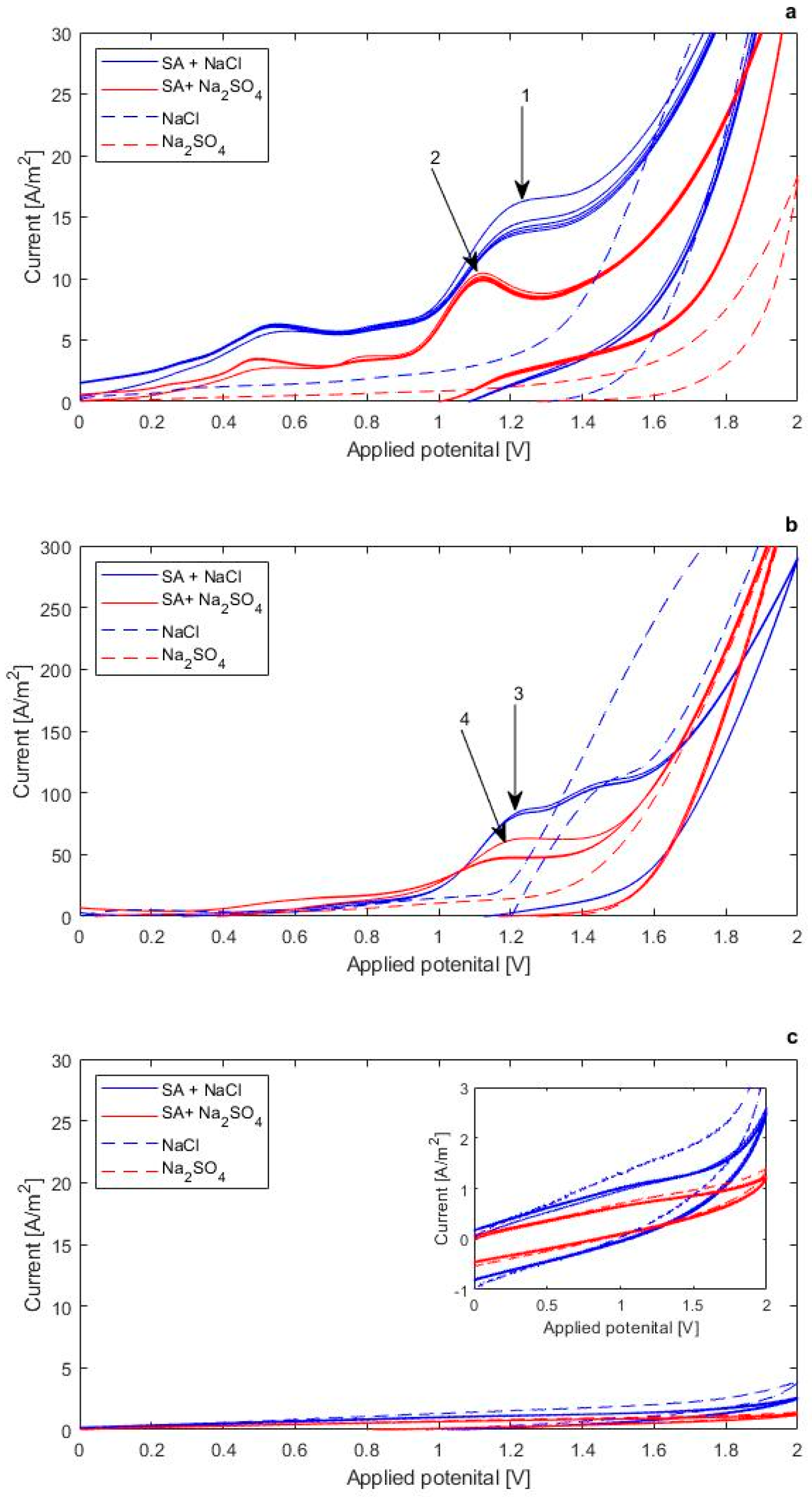
3.2.1. Electro-activity of Salicylic Acid
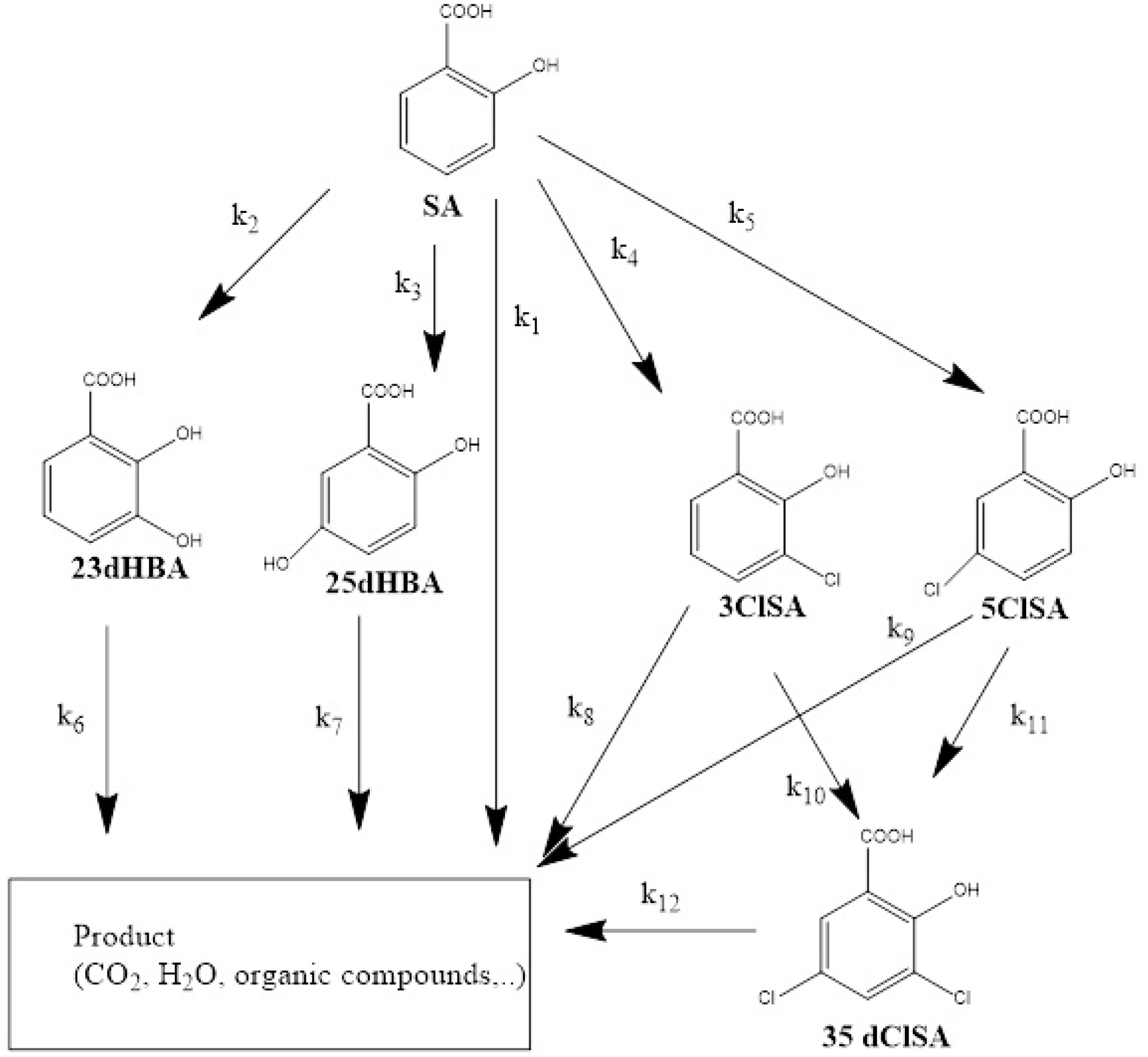
3.2.2. Formation of Hydroxylated Salicylic Acid Intermediates
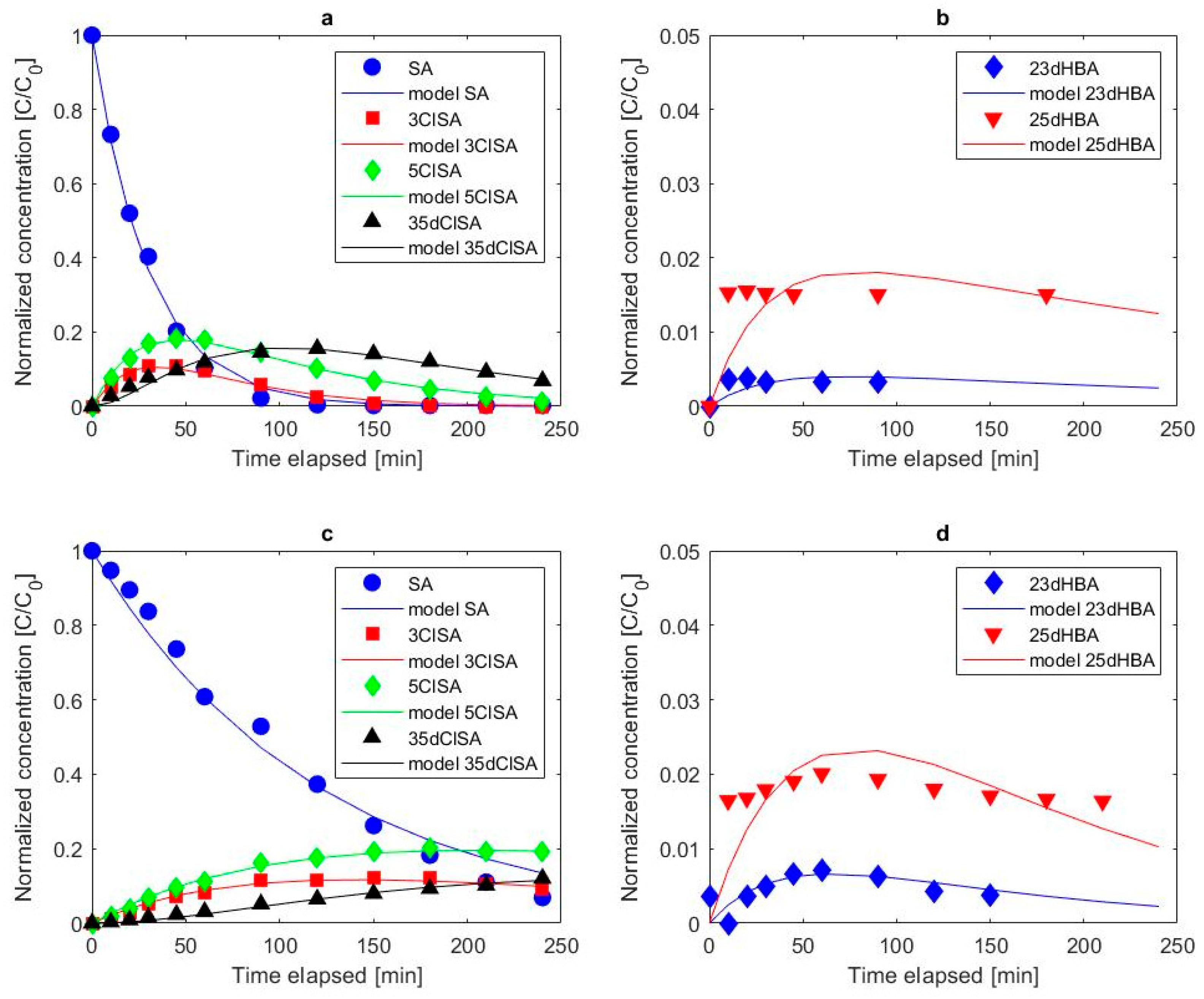
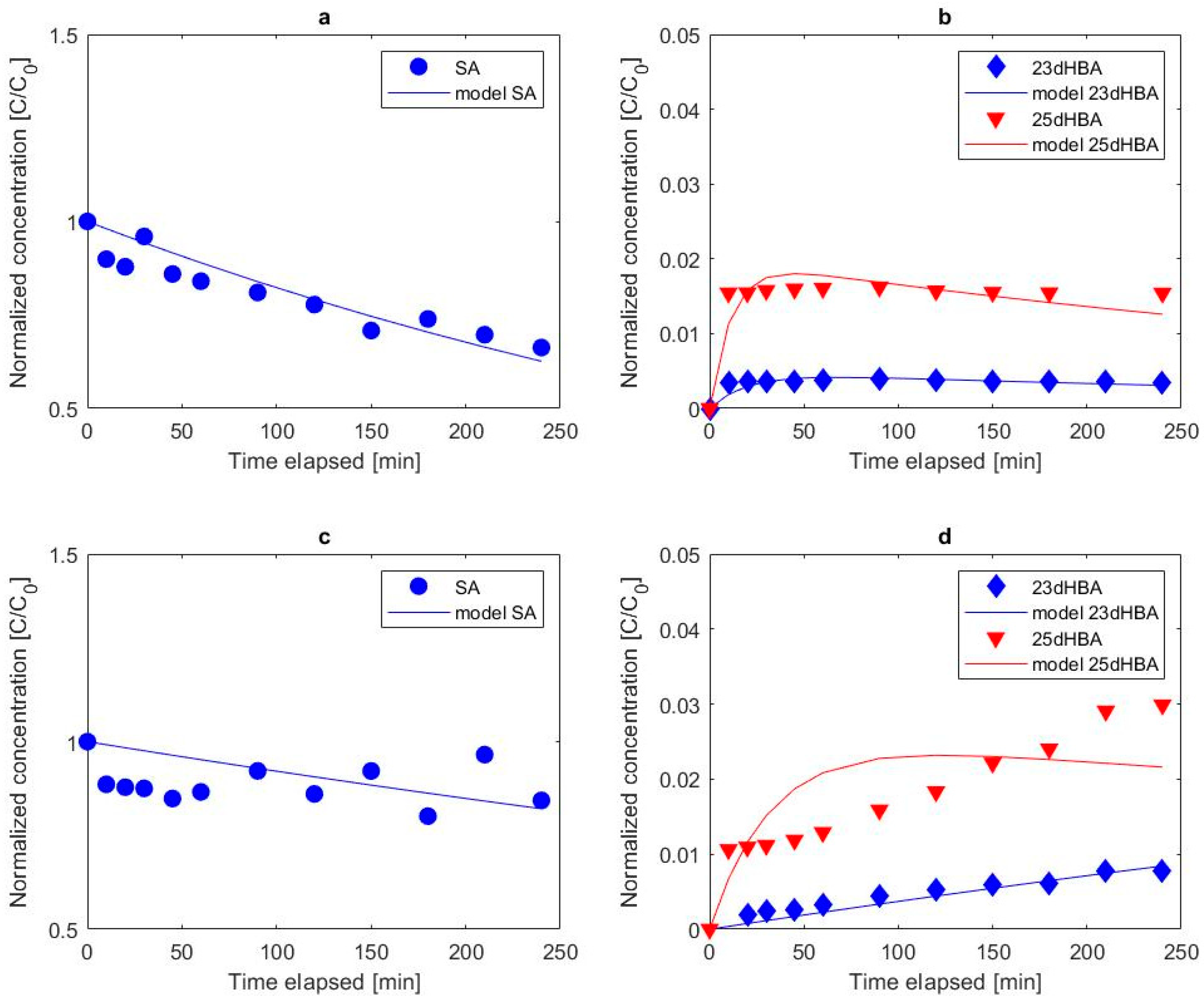
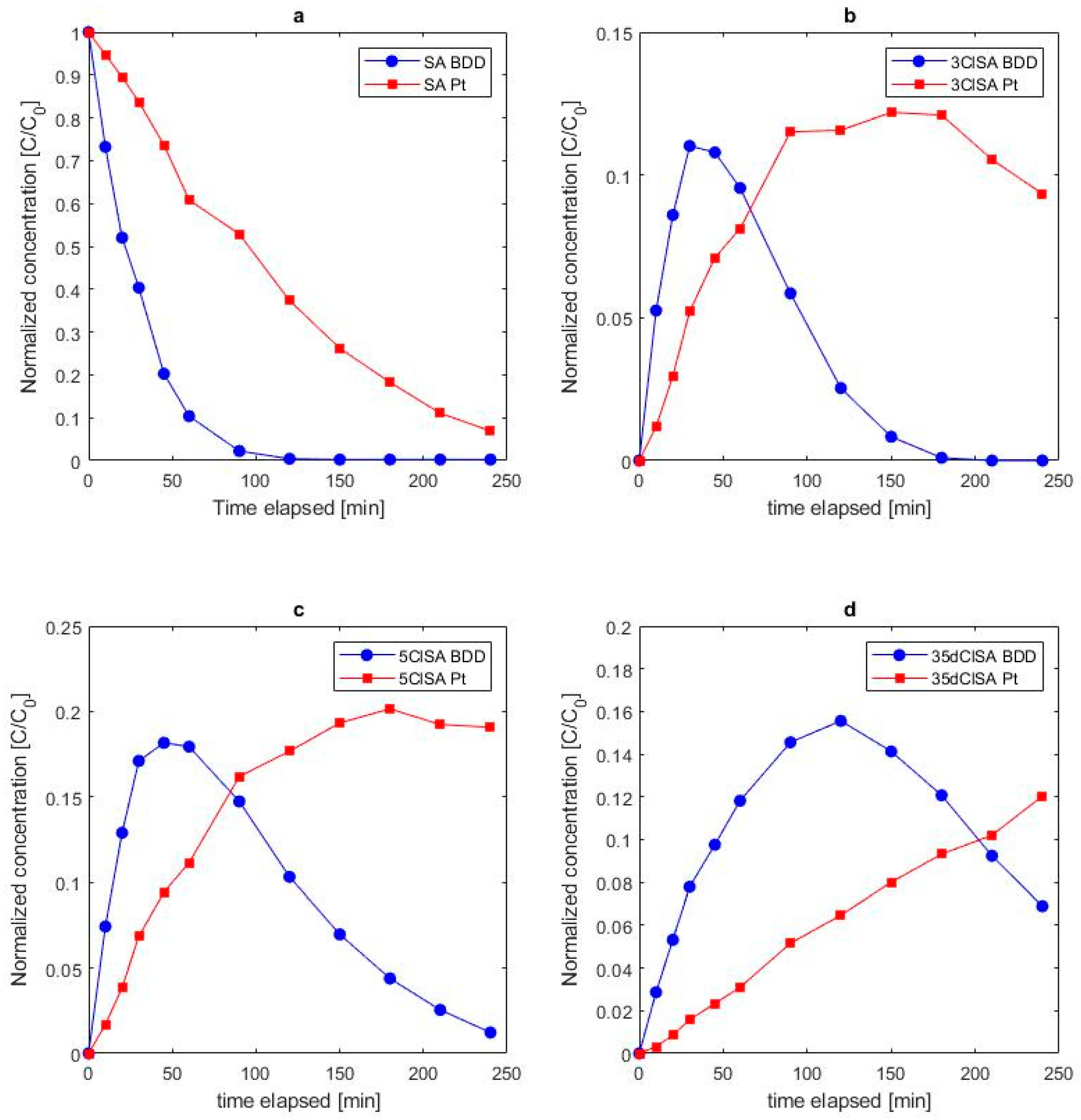
3.2.3. Formation of Chlorinated Salicylic Acid
3.2.4. Influence of Electrolyte Mediated Bulk Oxidation on Intermediate Formation
3.3. Overall Degradation Kinetics of Salicylic Acid
4. Conclusions
- This study confirms the formation of chlorinated intermediates. Three different chlorinated oxidation products were identified, 3ClSA, 5ClSA and 35dClSA, whereby 5ClSA was more frequently detected than 3ClSA and 35dClSA.
- Hydroxylation of salicylic acid via anodically generated hydroxyl radicals was confirmed via the identification and quantification of 23dHBA and 25dHBA. 25dHBA was more frequently formed than 23dHBA.
- Density functional theory and natural bond theory computations revealed the highest spin density at the C3 and C5 atom of salicylic acid. This explains the formation of the observed chlorinated and hydroxylated intermediates of salicylic acid, and why other intermediates like 26dHBA or 4ClSA were not detected.
- In chloride electrolyte, oxidation via mediating oxidizing species was found to be the governing oxidation process on both tested anode materials, whereas hydroxylation took place but at much lower rates than chlorination.
- Cyclic voltammetry confirmed of direct electron transfer of salicylic acid on Pt anodes, but not on BDD electrodes. The proposed kinetic model adequately describes the degradation of salicylic acid, and the formation of its chlorinated and hydroxylated intermediates and corresponding rate constants could be derived.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuoco, R.; Giannarelli, S. Integrity of aquatic ecosystems: An overview of a message from the South Pole on the level of persistent organic pollutants (POPs). Microchem. J. 2019, 148, 230–239. [Google Scholar] [CrossRef]
- Li, Z. Health risk characterization of maximum legal exposures for persistent organic pollutant (POP) pesticides in residential soil: An analysis. J. Environ. Manag. 2018, 205, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.C.; de Voogt, P. Persistent organic pollutants (POPs): State of the science. Environ. Pollut. 1999, 100, 209–221. [Google Scholar] [CrossRef]
- Wang, F.; Smith, D.W.; El-Din, M.G. Application of advanced oxidation methods for landfill leachate treatment–A review. J. Environ. Eng. Sci. 2003, 2, 413–427. [Google Scholar] [CrossRef]
- Panizza, M.; Delucchi, M.; Sirés, I. Electrochemical process for the treatment of landfill leachate. J. Appl. Electrochem. 2010, 40, 1721–1727. [Google Scholar] [CrossRef]
- Andreozzi, R.; Caprio, V.; Insola, A.; Marotta, R. Advanced oxidation processes (AOP) for water purification and recovery. Catal. Today 1999, 53, 51–59. [Google Scholar] [CrossRef]
- Dewil, R.; Mantzavinos, D.; Poulios, I.; Rodrigo, M.A. New perspectives for Advanced Oxidation Processes. J. Environ. Manag. 2017, 195, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Comninellis, C.; Chen, G. Electrochemistry for the Environment; Springer: New York, NY, USA, 2010. [Google Scholar] [CrossRef]
- Foti, G.; Gandini, D.; Comninellis, C.; Perret, A.; Haenni, W. Oxidation of organics by intermediates of water discharge on IrO2 and synthetic diamond anodes. Electrochem. Solid State Lett. 1999, 2. [Google Scholar] [CrossRef]
- Martínez-Huitle, C.A.; Ferro, S. Electrochemical oxidation of organic pollutants for the wastewater treatment: Direct and indirect processes. Chem. Soc. Rev. 2006, 35, 1324–1340. [Google Scholar] [CrossRef]
- Bonfatti, F.; Ferro, S.; Lavezzo, F.; Malacarne, M.; Lodi, G.; de Battisti, A. Electrochemical Incineration of Glucose as a Model Organic Substrate II. Role of Active Chlorine Mediation. J. Electrochem. Soc. 2000, 147, 592–596. [Google Scholar] [CrossRef]
- Stucki, S.; Kötz, R.; Carcer, B.; Suter, W. Electrochemical waste water treatment using high overvoltage anodes Part II: Anode performance and applications. J. Appl. Electrochem. 1991, 21, 99–104. [Google Scholar] [CrossRef]
- Iniesta, J.; Michaud, P.; Panizza, M.; Cerisola, G.; Aldaz, A.; Comninellis, C. Electrochemical oxidation of phenol at boron-doped diamond electrode. Electrochim. Acta 2001, 46, 3573–3578. [Google Scholar] [CrossRef]
- Guinea, E.; Arias, C.; Cabot, P.L.; Garrido, J.A.; Rodríguez, R.M.; Centellas, F.; Brillas, E. Mineralization of salicylic acid in acidic aqueous medium by electrochemical advanced oxidation processes using platinum and boron-doped diamond as anode and cathodically generated hydrogen peroxide. Water Res. 2008, 42, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; van Hullebusch, E.D.; Rodrigo, M.A.; Esposito, G.; Oturan, M.A. Removal of residual anti-inflammatory and analgesic pharmaceuticals from aqueous systems by electrochemical advanced oxidation processes, A. review. Chem. Eng. J. 2013, 228, 944–964. [Google Scholar] [CrossRef]
- Farinholt, L.H.; Stuart, A.P.; Twiss, D. The Halogenation of Salicylic Acid. J. Am. Chem. Soc. 1940, 62, 1237–1241. [Google Scholar] [CrossRef]
- Broadwater, M.A.; Swanson, T.L.; Sivey, J.D. Emerging investigators series: Comparing the inherent reactivity of often-overlooked aqueous chlorinating and brominating agents toward salicylic acid. Environ. Sci. Water Res. Technol. 2018, 4, 369–384. [Google Scholar] [CrossRef]
- Torriero, A.A.J.; Luco, J.M.; Sereno, L.; Raba, J. Voltammetric determination of salicylic acid in pharmaceuticals formulations of acetylsalicylic acid. Talanta 2004, 62, 247–254. [Google Scholar] [CrossRef]
- Wudarska, E.; Chrzescijanska, E.; Kusmierek, E. Electroreduction of Salicylic Acid, Acetylsalicylic Acid and Pharmaceutical Products Containing these Compounds. Port. Electrochim. Acta 2014, 32, 295–302. [Google Scholar] [CrossRef]
- Wragg, A.A.; Tagg, D.J.; Patrick, M.A. Diffusion-controlled current distributions near cell entries and corners. J. Appl. Electrochem. 1980, 10, 43–47. [Google Scholar] [CrossRef]
- Chatzisymeon, E.; Xekoukoulotakis, N.P.; Diamadopoulos, E.; Katsaounis, A.; Mantzavinos, D. Boron-doped diamond anodic treatment of olive mill wastewaters: Statistical analysis, kinetic modeling and biodegradability. Water Res. 2009, 43, 3999–4009. [Google Scholar] [CrossRef] [Green Version]
- Panizza, M.; Kapalka, A.; Comninellis, C. Oxidation of organic pollutants on BDD anodes using modulated current electrolysis. Electrochim. Acta 2008, 53, 2289–2295. [Google Scholar] [CrossRef]
- Frisch, D.J.F.M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision B.01. 2010. Available online: http://gaussian.com/ (accessed on 13 October 2018).
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Weigend, F.; Furche, F.; Ahlrichs, R. Gaussian basis sets of quadruple zeta valence quality for atoms H-Kr. J. Chem. Phys. 2003, 119, 12753–12762. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H. to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Jing, Y.; Chaplin, B.P. Mechanistic Study of the Validity of Using Hydroxyl Radical Probes To Characterize Electrochemical Advanced Oxidation Processes. Environ. Sci. Technol. 2017, 51, 2355–2365. [Google Scholar] [CrossRef]
- Evans, D.; Hart, J.P.; Rees, G. Voltammetric Behaviour of Salicylic Acid at a Glassy Carbon Electrode and Its Determination in Serum Using Liquid Chromatography With Amperometric Detection. Analyst 1991, 116, 803–806. [Google Scholar] [CrossRef]
- Dubois, D.; Moninot, G.; Kutner, W.; Jones, M.T.; Kadish, K.M. Electroreduction of Buckmlnsterfullerene, Electrolyte, and Temperature Effects Aprotic Solvents: Solvent Supporting. J. Phys. Chem. 1992, 7137–7145. [Google Scholar] [CrossRef]
- Lee, J.H.Q.; Koh, Y.R.; Webster, R.D. The electrochemical oxidation of diethylstilbestrol (DES) in acetonitrile. J. Electroanal. Chem. 2017, 799, 92–101. [Google Scholar] [CrossRef]
- Louhichi, B.; Bensalash, N.; Gadri, A. Electrochemical oxidation of benzoic acid derivatives on boron doped diamond: Voltammetric study and galvanostatic electrolyses. Chem. Eng. Technol. 2006, 29, 944–950. [Google Scholar] [CrossRef]
- Montilla, F.; Michaud, P.A.; Morallon, E.; Vazquez, J.L.; Comninellis, C.; Morallón, E.; Vázquez, J.L. Electrochemical oxidation of benzoic acid at boron-doped diamond electrodes. Electrochim. Acta 2002, 47, 3509–3513. [Google Scholar] [CrossRef]
- Madsen, H.T.; Søgaard, E.G.; Muff, J. Chemosphere Study of degradation intermediates formed during electrochemical oxidation of pesticide residue 2,6-dichlorobenzamide (BAM) at boron doped diamond (BDD) and platinum–iridium anodes. Chemosphere 2014, 109, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Farhat, A.; Keller, J.; Tait, S.; Radjenovic, J. Removal of Persistent Organic Contaminants by Electrochemically Activated Sulfate. Environ. Sci. Technol. 2015, 49. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position Hydroxylate Product | Relative Energy (kJ/mol) | Position Mono-Chlorinated Product | Relative Energy (kJ/mol) | Position Di-Chlorinated product | Relative Energy (kJ/mol) |
|---|---|---|---|---|---|
| 4 | 0.0 | 4 | 0.0 | 4,5 | 0.0 |
| 6 | 10.8 | 5 | 3.9 | 3,5 | 0.1 |
| 5 | 15.7 | 3 | 9.7 | 3,4 | 4.8 |
| 3 | 18.2 | 6 | 29.6 | 4,6 | 17.0 |
| n/a | n/a | n/a | n/a | 3,6 | 25.5 |
| n/a | n/a | n/a | n/a | 5,6 | 32.6 |
| Anode | GC | Pt | BDD | |||
|---|---|---|---|---|---|---|
| Electrolyte | NaCl | Na2SO4 | NaCl | Na2SO4 | NaCl | Na2SO4 |
| Epa (V) SA | 1.20 | 1.07 | 1.22 | 1.15 | n/a | n/a |
| Rate Constants (1/min) | NaCl/BDD | NaCl/Pt | Na2SO4/BDD | Na2SO4/Pt |
|---|---|---|---|---|
| k1 | 1.28 × 10−2 | 2.24 × 10−3 | 3.32× 10−14 | 2.22 × 10−14 |
| k2 | 1.75 × 10−4 | 2.89 × 10−4 | 2.41× 10−4 | 3.87 × 10−5 |
| k3 | 7.62 × 10−4 | 8.17 × 10−4 | 1.71 × 10−3 | 7.82 × 10−4 |
| k4 | 8.56 × 10−3 | 2.27 × 10−3 | n/a | n/a |
| k5 | 1.10 × 10−2 | 2.75 × 10−3 | n/a | n/a |
| k6 | 3.63 × 10−3 | 2.53 × 10−2 | 5.07 × 10−2 | 3.20 × 10−13 |
| k7 | 2.89 × 10−3 | 1.80 × 10−2 | 8.70 × 10−2 | 3.05× 10−2 |
| k8 | 2.22 × 10−14 | 2.22× 10−14 | n/a | n/a |
| k9 | 9.81 × 10−3 | 2.22 × 10−14 | n/a | n/a |
| k10 | 2.59 × 10−2 | 5.96 × 10−3 | n/a | n/a |
| k11 | 3.55 × 10−3 | 2.53 × 10−3 | n/a | n/a |
| k12 | 9.96 × 10−3 | 7.71 × 10−3 | n/a | n/a |
| kSA, Σ(k1–k5) | 3.33 × 10−2 | 8.37 × 10−3 | 1.95 × 10−3 | 8.21 × 10−4 |
| Fitting Quality, R2 (%) | ||||
| SA | 98% | 96% | 95% | 92% |
| 23dHBA | 77% | 88% | 87% | 88% |
| 25dHBA | 78% | 80% | 88% | 64% |
| 3ClSA | 94% | 95% | n/a | n/a |
| 5ClSA | 96% | 97% | n/a | n/a |
| 35dClSA | 93% | 96% | n/a | n/a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambauen, N.; Muff, J.; Mai, N.L.; Hallé, C.; Trinh, T.T.; Meyn, T. Insights into the Kinetics of Intermediate Formation during Electrochemical Oxidation of the Organic Model Pollutant Salicylic Acid in Chloride Electrolyte. Water 2019, 11, 1322. https://doi.org/10.3390/w11071322
Ambauen N, Muff J, Mai NL, Hallé C, Trinh TT, Meyn T. Insights into the Kinetics of Intermediate Formation during Electrochemical Oxidation of the Organic Model Pollutant Salicylic Acid in Chloride Electrolyte. Water. 2019; 11(7):1322. https://doi.org/10.3390/w11071322
Chicago/Turabian StyleAmbauen, Noëmi, Jens Muff, Ngoc Lan Mai, Cynthia Hallé, Thuat T. Trinh, and Thomas Meyn. 2019. "Insights into the Kinetics of Intermediate Formation during Electrochemical Oxidation of the Organic Model Pollutant Salicylic Acid in Chloride Electrolyte" Water 11, no. 7: 1322. https://doi.org/10.3390/w11071322