Stochastic Method for Evaluating Removal, Fate and Associated Uncertainties of Micropollutants in a Stormwater Biofilter at an Annual Scale

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Methods
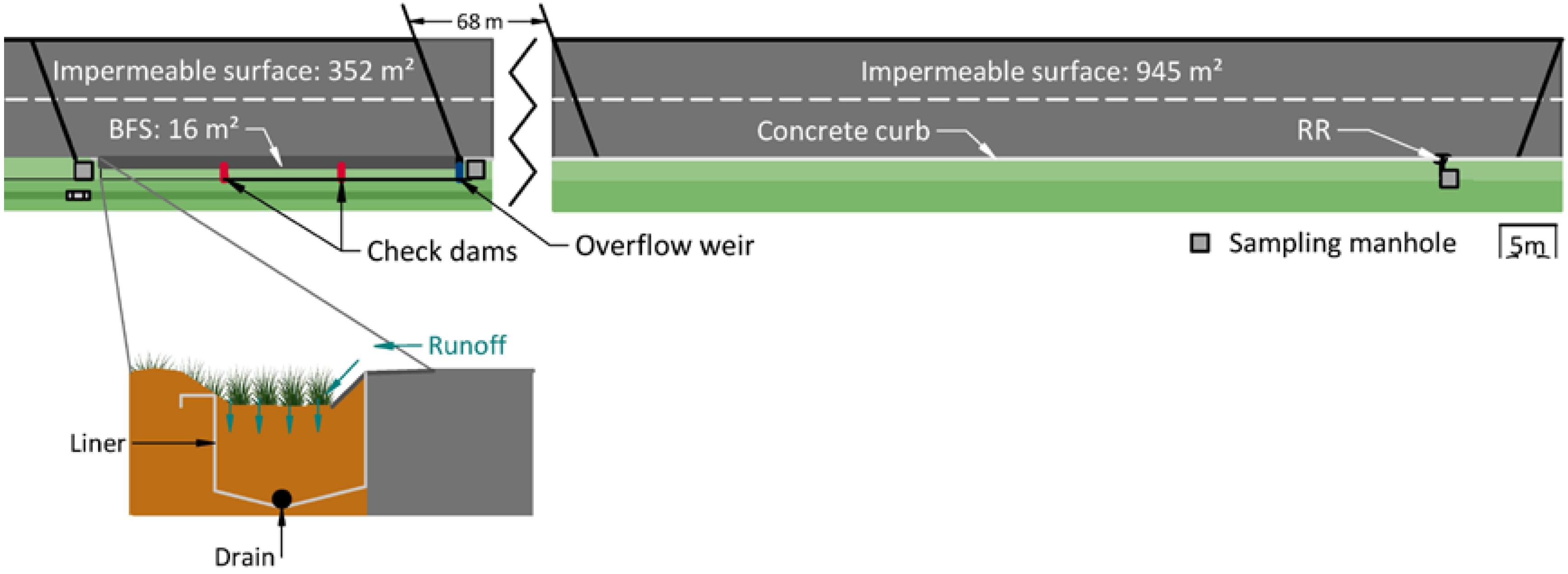
2.1.1. Site Description
2.1.2. Continuous Measurements
2.1.3. Water Sampling and Pre-Treatment
2.1.4. Soil Sampling and Preparation
2.1.5. Analytical Methods
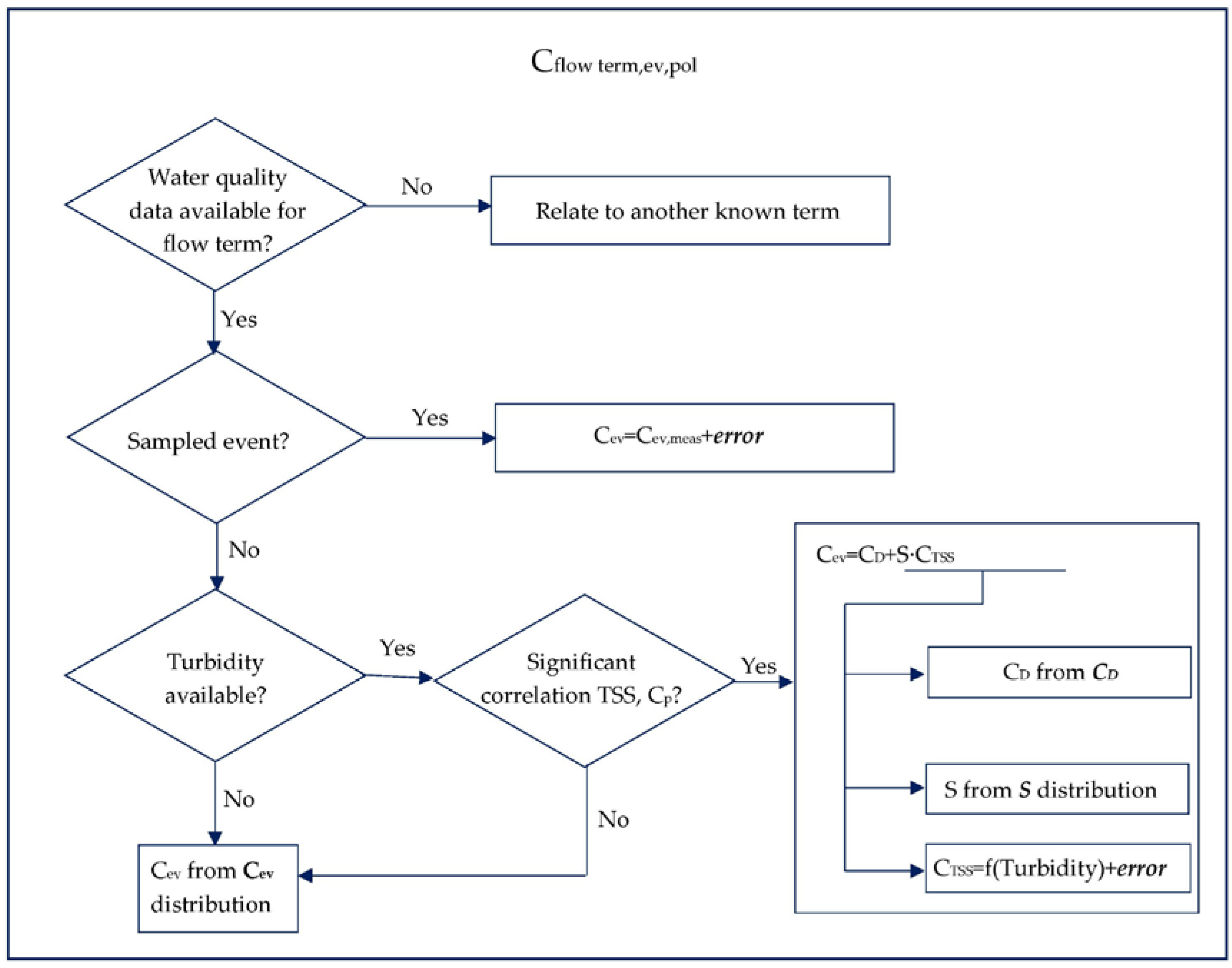
2.2. Mass Balance and Uncertainty Calculations
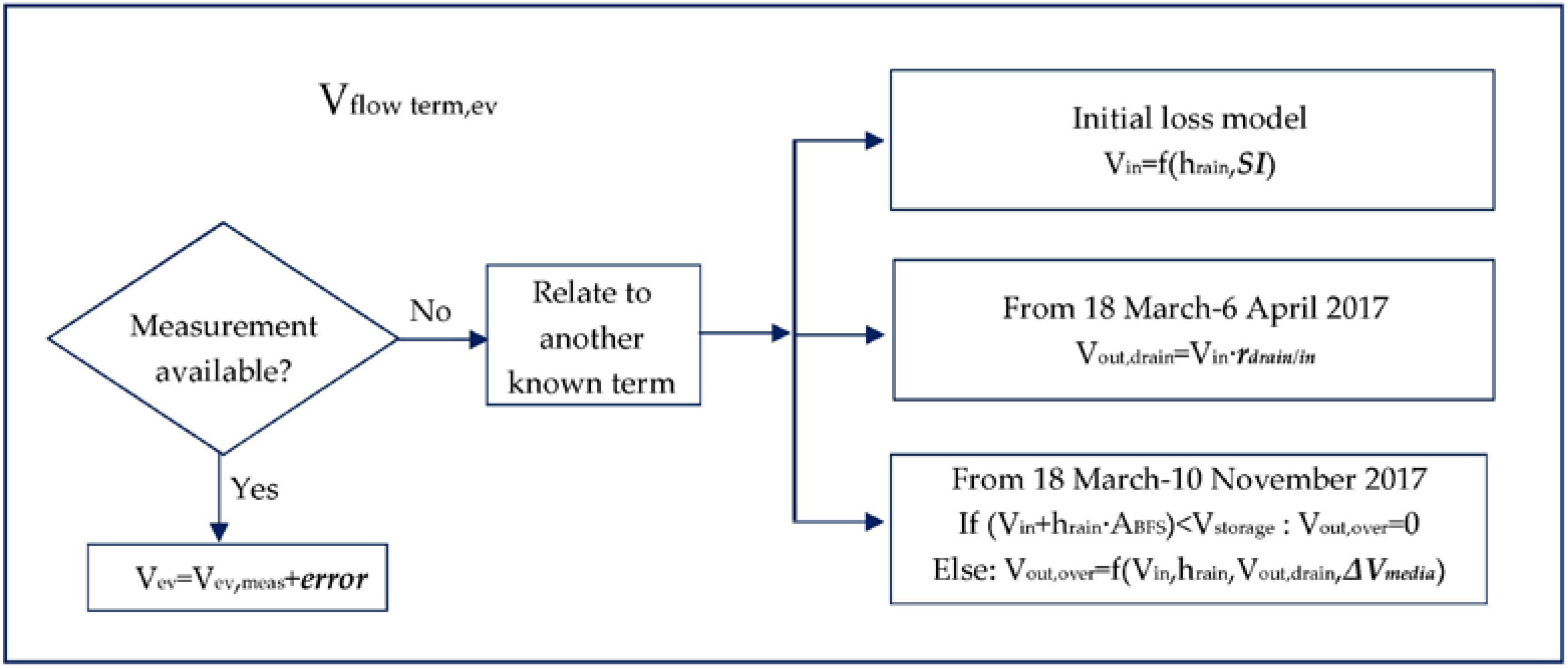
2.2.1. Loads Associated with Water Flows
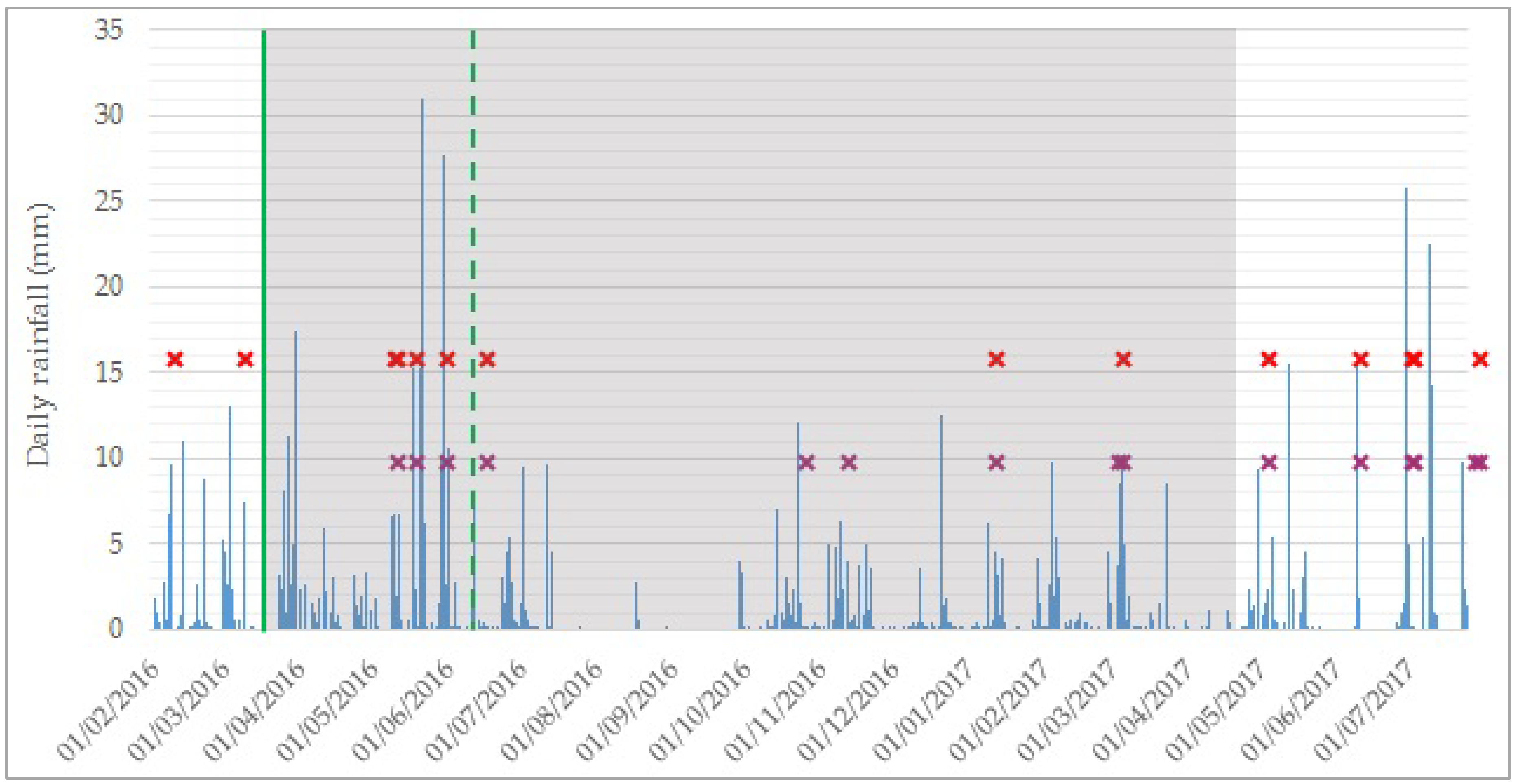
Separation of Rain Events
Min,RR
Mout,drain
Mout,over
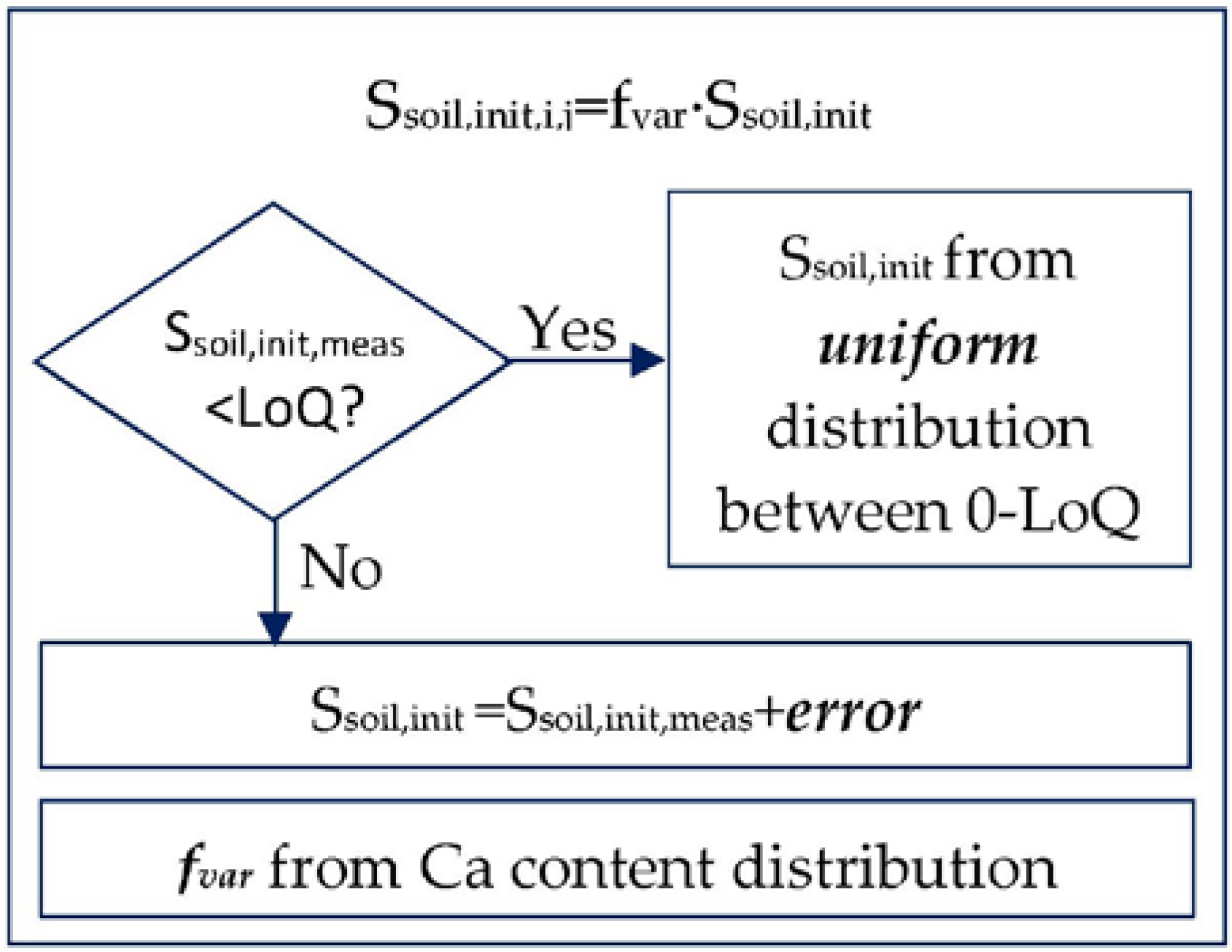
2.2.2. Mass Stored in Soil
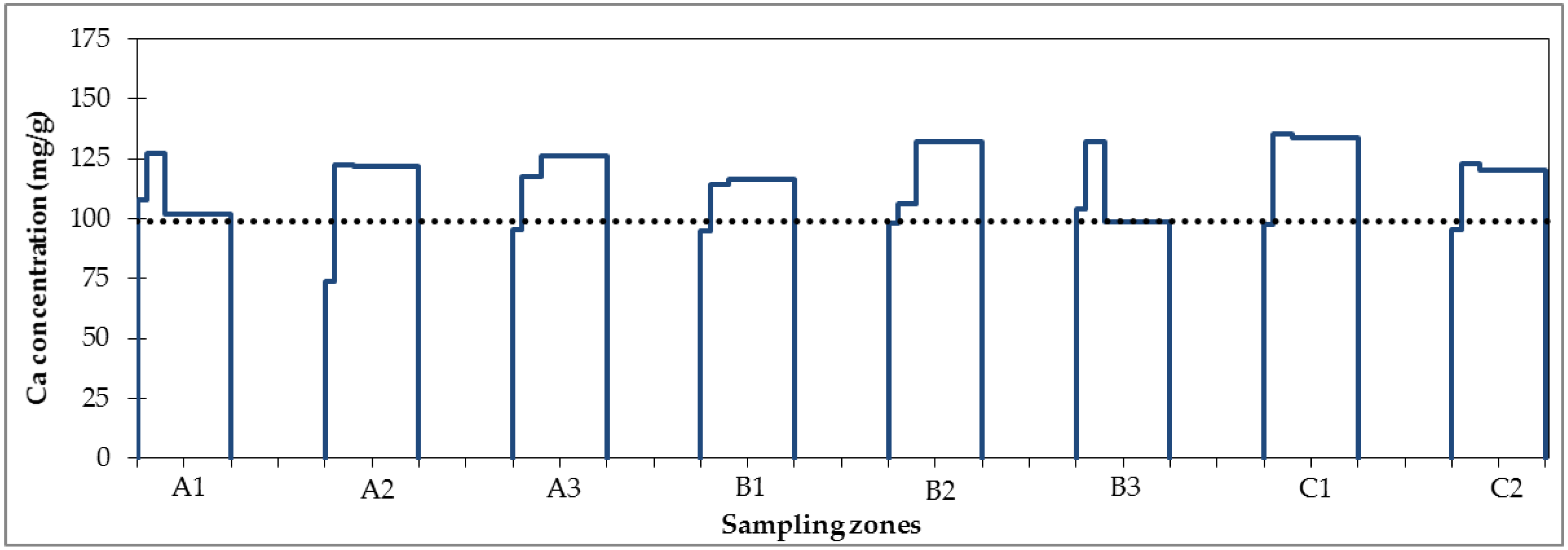
Definition of Soil Properties
Definition of Ssoil,init,i,j
3. Results and Discussion
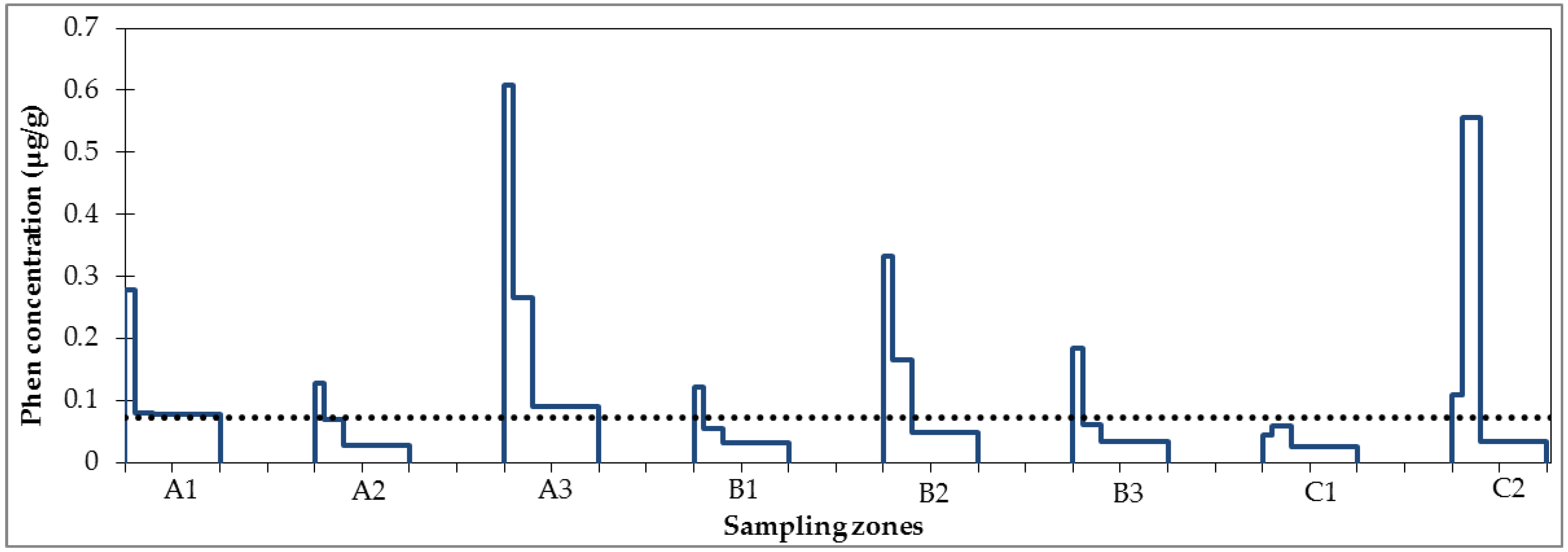
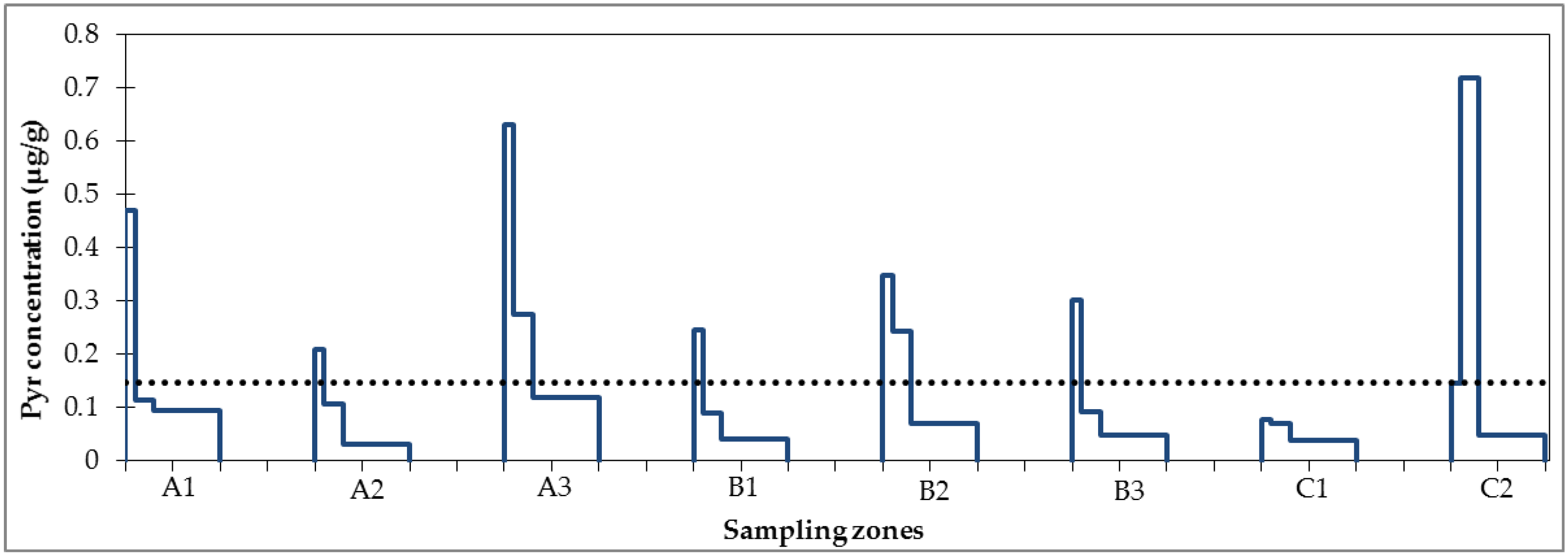
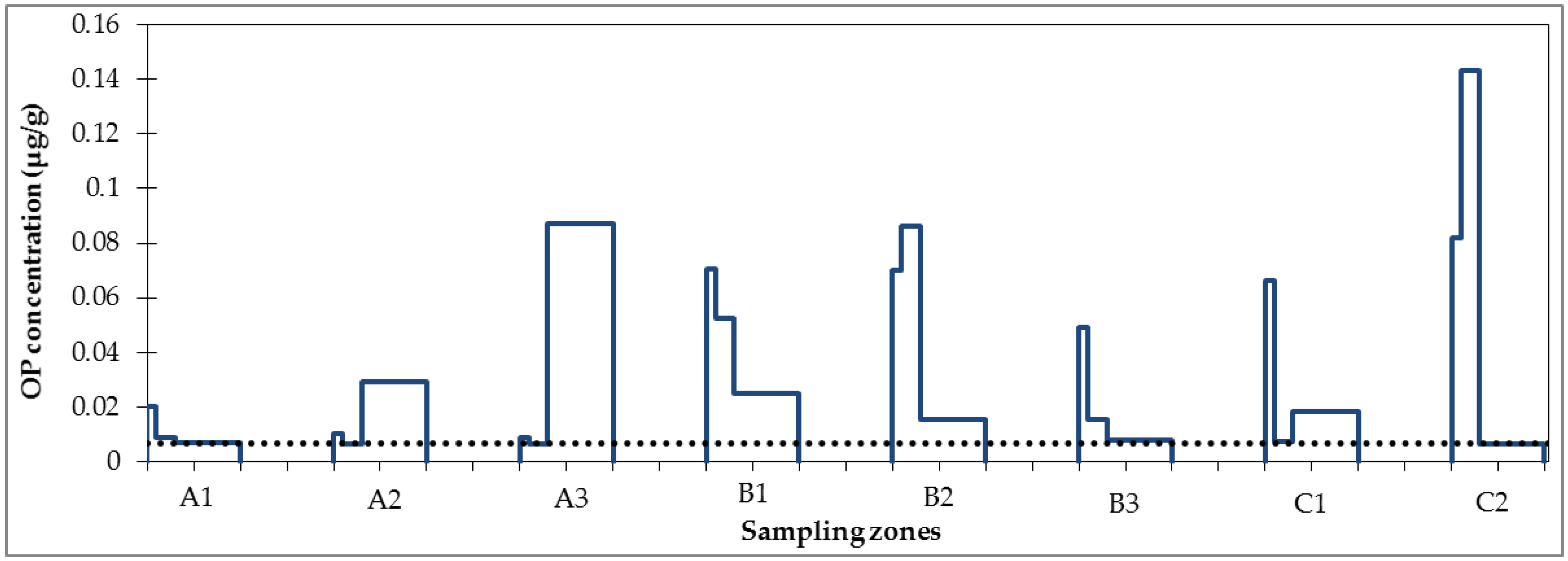
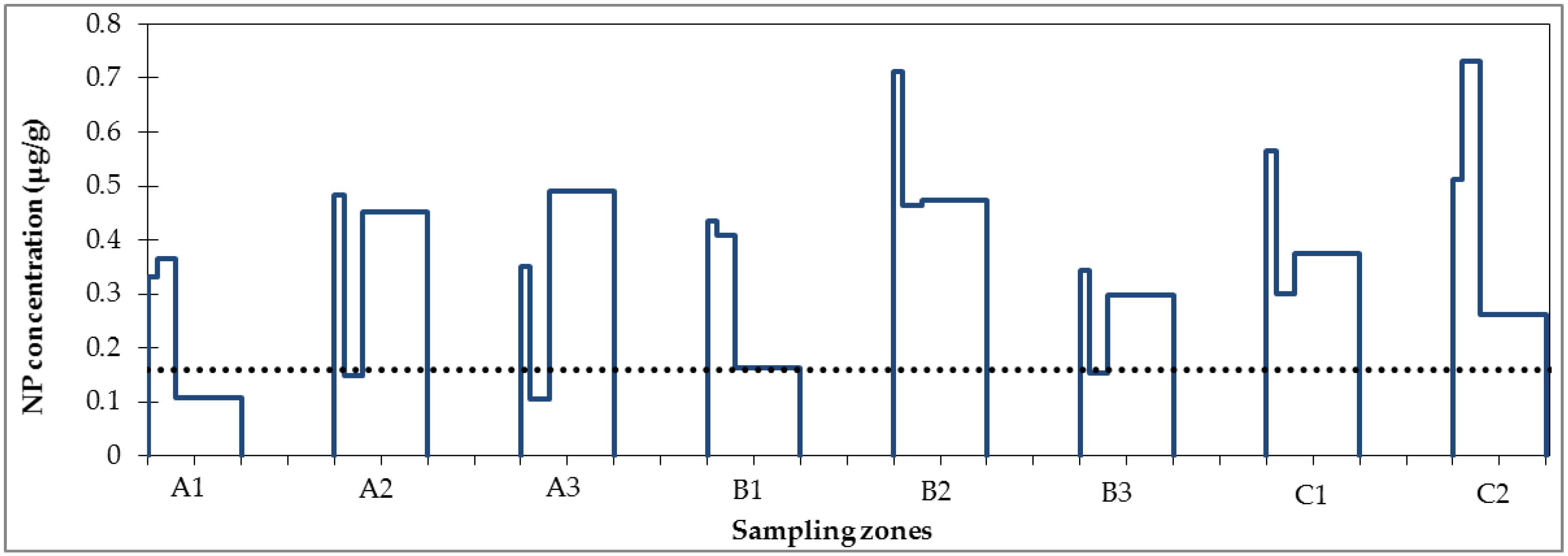
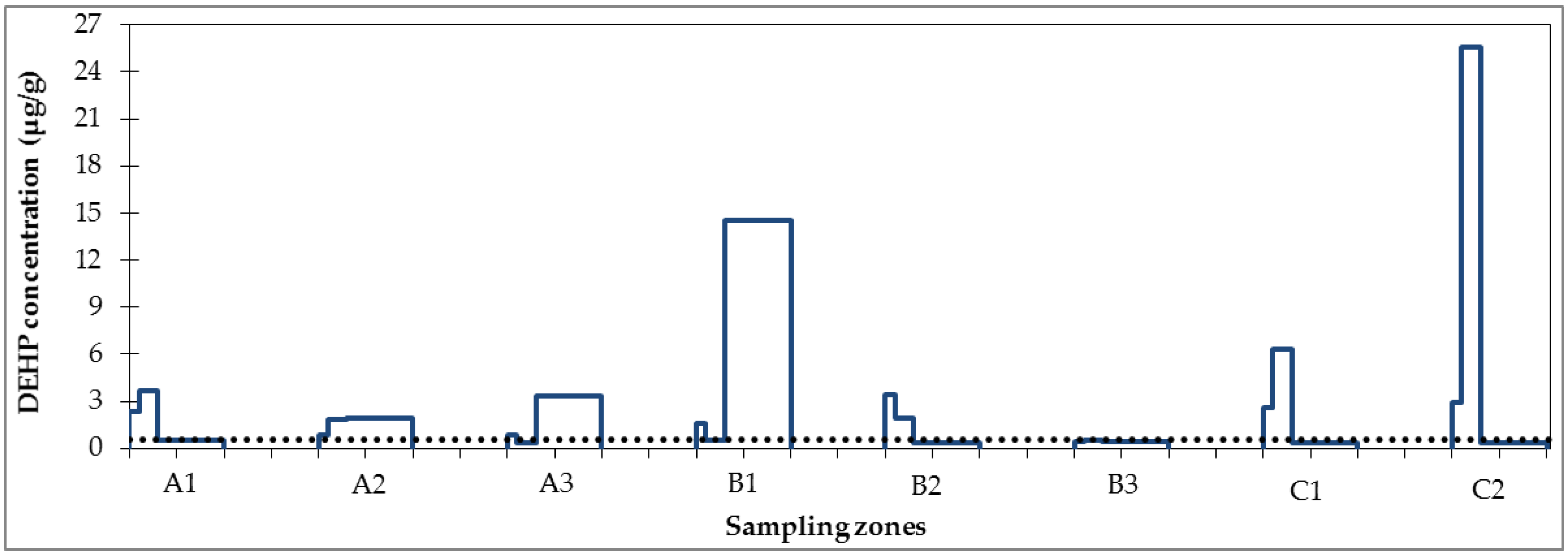
3.1. Micropollutant Load in Road Runoff
3.2. Micropollutant Load Reduction
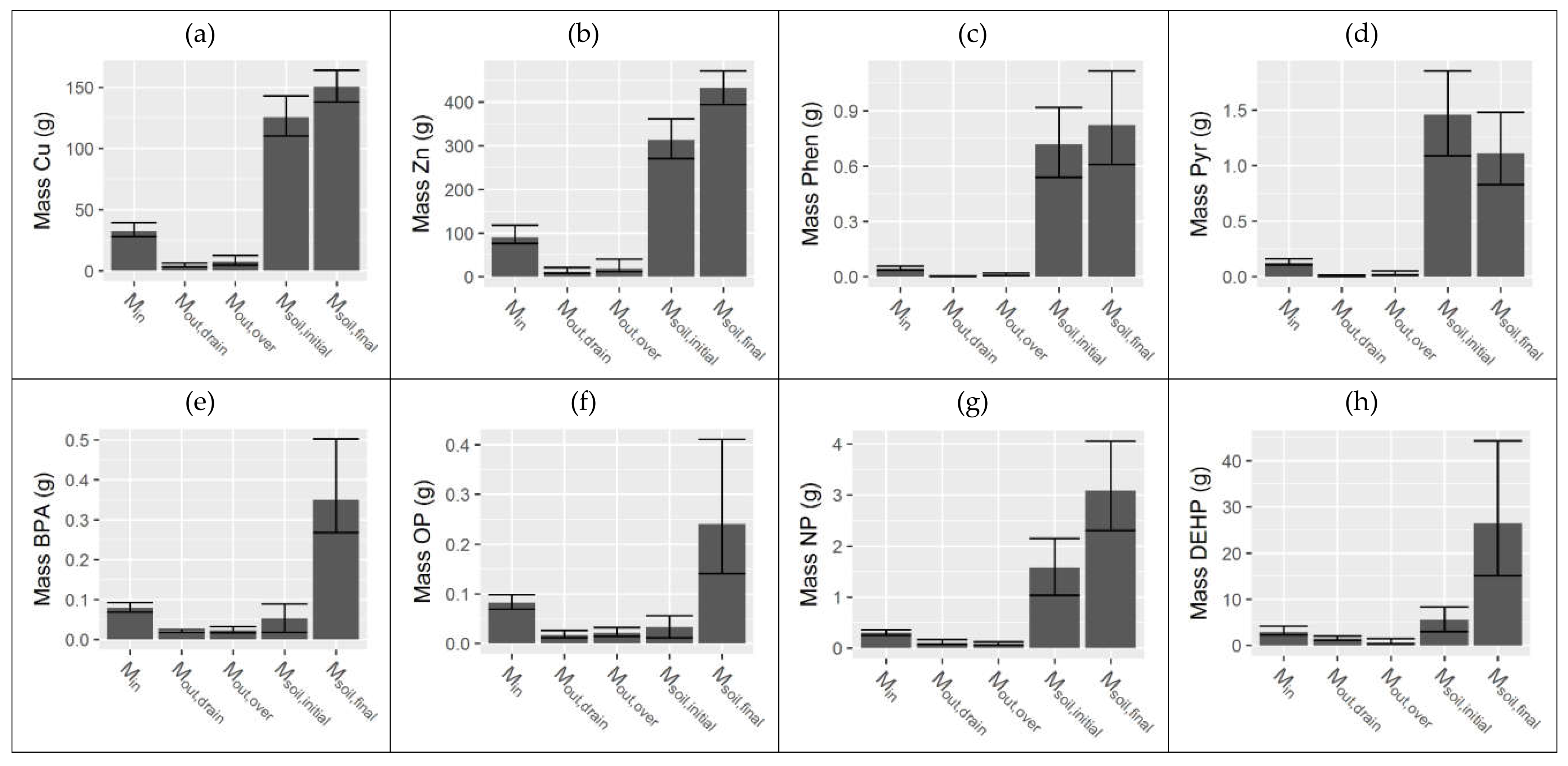
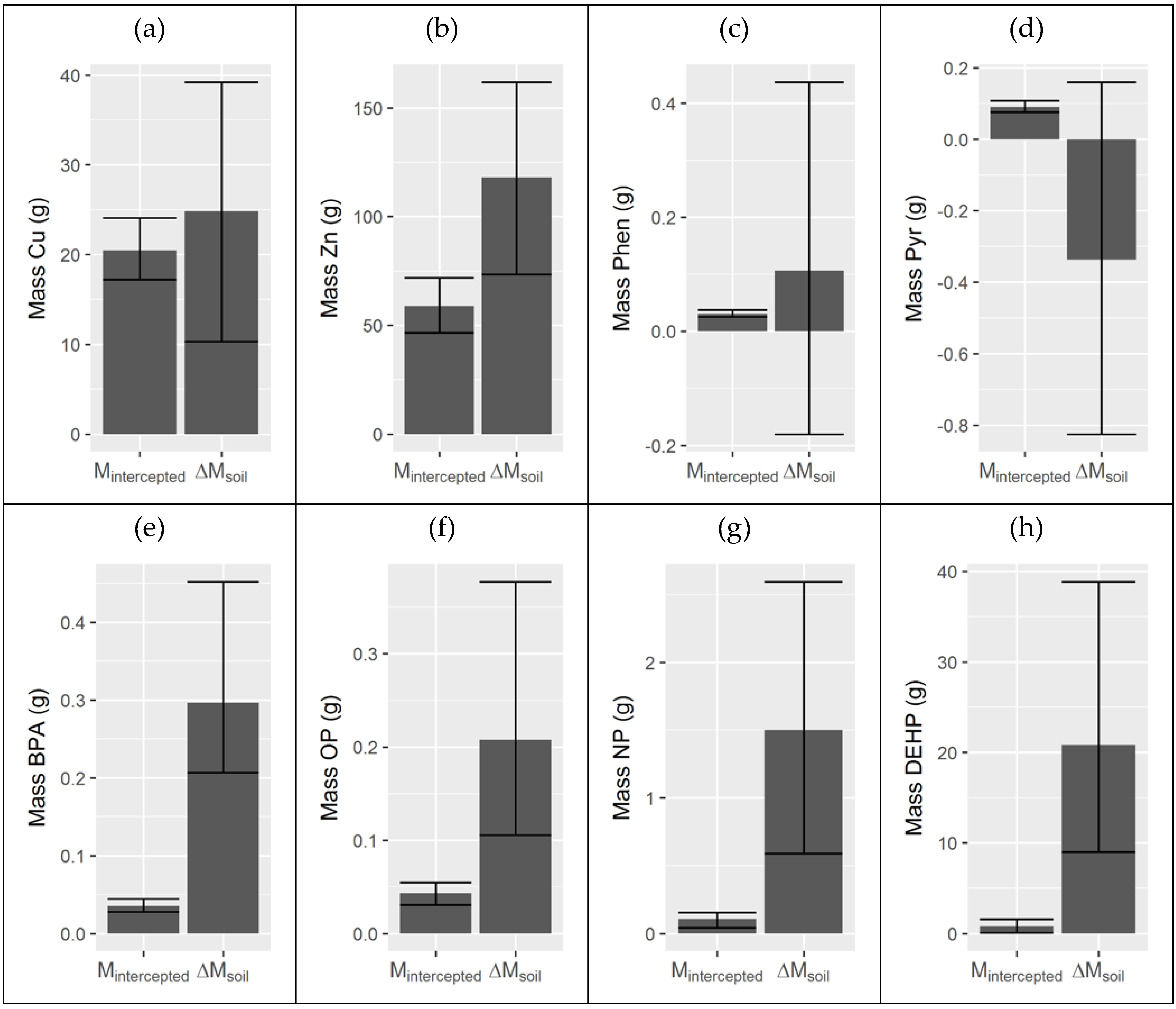
3.3. Integrated Water and Filter Media Mass Balance
3.4. Limitations of the Proposed Mass Balance Approach
3.4.1. Questionable Representativity of the Initial Soil Sample
3.4.2. Overestimation of Outlet Pollutant Loads
3.4.3. Atmospheric Deposition
3.4.4. Galvanized Steel Barrier
3.4.5. Degradation of Ethoxylated Alkylphenols
3.4.6. Biofiltration Swale Construction Materials
3.5. Necessary Conditions for a Mass Balance Significantly Demonstrating Dissipation
3.5.1. Pollutant Loads in and out of the System Have Been Evaluated, Accounting for Uncertainties, for all Important Pollutant Flux Terms
3.5.2. Both Initial and Final Concentrations in Soil Have Been Evaluated in a Representative Fashion; Soil Properties and Their Variability Have Been Characterized
3.5.3. No Pollutant Sources are Present within the System or the Dissipation of a Given Pollutant Exceeds Its Production/Emission (in the Second Case, Dissipation Will Be Underestimated but May Still Be Identified)
3.5.4. Intercepted Pollutant Masses Are Significant Compared to Initial Soil Pollutant Mass
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Daily Rainfall and Timeline of Sampled Events

Appendix B. Distributions Used for Stochastic Components of the Mass Balance Calculation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pollutant | Distribution Analytical Error |
|---|---|
| TSS | Normal, μ = 0, σ = 2.9% |
| Cu | Diss: Normal, μ = 0, σ = 5% Part: Normal, μ = 0, σ = 7% |
| Zn | Diss: Normal, μ = 0, σ = 7% Part: Normal, μ = 0, σ = 7% |
| Pyr | Diss: Normal, μ = 0, σ = 20% Part: Normal, μ = 0, σ = 10% |
| Phen | Diss: Normal, μ = 0, σ = 20% Part: Normal, μ = 0, σ = 10% |
| BPA | Diss: Normal, μ = 0, σ = 22% Part: Normal, μ = 0, σ = 20% |
| OP | Diss: Normal, μ = 0, σ = 48% Part: Normal, μ = 0, σ = 37% |
| NP | Diss: Normal, μ = 0, σ = 29% Part: Normal, μ = 0, σ = 34% |
| DEHP | Diss: Normal, μ = 0, σ = 41% Part: Normal, μ = 0, σ = 30% |
| Term | Case | Stochastic Component | Distribution Type and Parameters |
|---|---|---|---|
| Vin | Always | Initial loss | Uniform, 0.4–1 mm |
| Vdrain | Measurement available | Error V | Normal, μ = 0, σ = 5% |
| Vdrain | No measurement available | Fraction of Vin drained | Log-normal, μln = −2.04, σln = 1.22 |
| Vover | Measurement available | Error h | Normal, μ = 0, σ = 0.5% |
| Vover | No measurement available | ΔVmedia,ev | Uniform, 0–1240 L |
| CTSS,in | Turbidity < 50 FNU | CTSS,in | Uniform, 0–70 mg/L |
| CTSS,in | Valid turbidity measurement | Regression error | Normal, μ = 0, σ = 71 mg/L |
| CTSS,in | No turbidity measurement | CTSS,in | Log-normal, μln = 5.44, σln = 0.76 mg/L |
| CTSS,drain | Winter events | Relationship to CTSS,in | Uniform, 0.225–0.883 |
| CTSS,drain | Events outside winter | CTSS,drain | Log-normal, μln = 2.92, σln = 0.59 mg/L |
| Cev | Sampled events | Analytical error | Normal, μ = 0, σ = σuncertainty, pollutant |
| Pollutant | Cev Distribution (µg/L) | CD,ev Distribution (µg/L) | Sev Distribution (µg/g) |
|---|---|---|---|
| Cu | Log-normal μln = 5.39, σln = 0.52 | Log-normal μln = 5.39, σln=0.52 | Log-normal μln = 6.41, σln = 0.18 |
| Zn | Log-normal μln = 6.37, σln = 0.65 | Log-normal μln = 6.37, σln = 0.65 | Log-normal μln = 7.48, σln = 0.19 |
| Pyr | Log-normal μln = −0.26, σln = 0.66 | Log-normal μln = −3.84, σln = 0.27 | Log-normal μln = 0.99, σln = 0.24 |
| Phen | Log-normal μln = −1.41, σln = 0.74 | Log-normal μln = −3.62, σln = 0.69 | Log-normal μln = −0.16, σln = 0.38 |
| BPA | Log-normal μln = −0.77, σln = 0.41 | Log-normal μln = −1.21, σln = 0.59 | Log-normal μln = −0.75, σln = 0.52 |
| OP | Log-normal μln = −0.74, σln = 0.49 | Normal μln = 0.14, σln = 0.05 | Log-normal μln = 0.16, σln = 0.42 |
| NP | Log-normal μln = 0.52, σln = 0.48 | - | - |
| DEHP | Log-normal μln = 2.76, σln = 0.83 | Log-normal μln = 1.12, σln = 0.68 | Log-normal μln = 3.63, σln = 0.76 |
| Pollutant | Cev Distribution (µg/L) | %Below LoQ | Distribution when < LoQ (µg/L) | Distribution when > LoQ (µg/L) |
|---|---|---|---|---|
| Cu | Log-normal μln = 3.65, σln = 0.94 | - | - | - |
| Zn | Log-normal μln = 4.05, σln = 1.39 | - | - | - |
| Pyr | - | CD,ev: 30% CP,ev: 0% | CD,ev: Uniform 0–0.01 | CD,ev: Log-normal μln = −3.80, σln = 0.43 Part: Log-normal μln = −3.09, σln = 1.43 |
| Phen | - | CD,ev: 69% CP,ev: 15% | CD,ev: Uniform 0–0.01 CP,ev Uniform 0–0.004 | CD,ev: Unform 0.012–0.098 CP,ev: Log-normal μln = −3.88, σln = 1.29 |
| BPA | Log-normal μln = −1.43, σln = 0.50 | - | - | - |
| OP | Log-normal μln = −2.19, σln = 1.14 | - | - | - |
| NP | Log-normal μln = −0.16, σln = 1.09 | - | - | - |
| DEHP | Log-normal μln = 2.66, σln = 0.85 | - | - | - |
| Term | Case | Stochastic Component | Distribution Type and Parameters |
|---|---|---|---|
| Ssamp | <LoQ | Ssamp | Uniform 0-LoQ |
| Ssamp | >LoQ | Analytical Error | Normal, μ = 0, σ = σuncertainty, pollutant |
| fsample | Zone hypothesis valid | Zone error | Zone 1: a = Uniform, −0.1–0.1 Zone 2: if a < 0: b = Uniform, (0.1-a)–0.1 else b = Uniform, −0.1-(0.1-a) Zone 3: c = −(a + b) |
| fsample | Zone hypothesis invalid | fsample | Zone 1: a = Uniform, 0–1 Zone 2: b = Uniform, 0-(1-a) Zone 3: c = 1-a-b |
| f<2mm | Always | f<2mm | Normal, μ = 0.733, σ = 0.045 |
| ρ | Always | Variability at depth | Uniform, −0.2–0.2 |
| Sinit | Always | Initial composition error | Normal, μ = 0, σ = 0.139 |
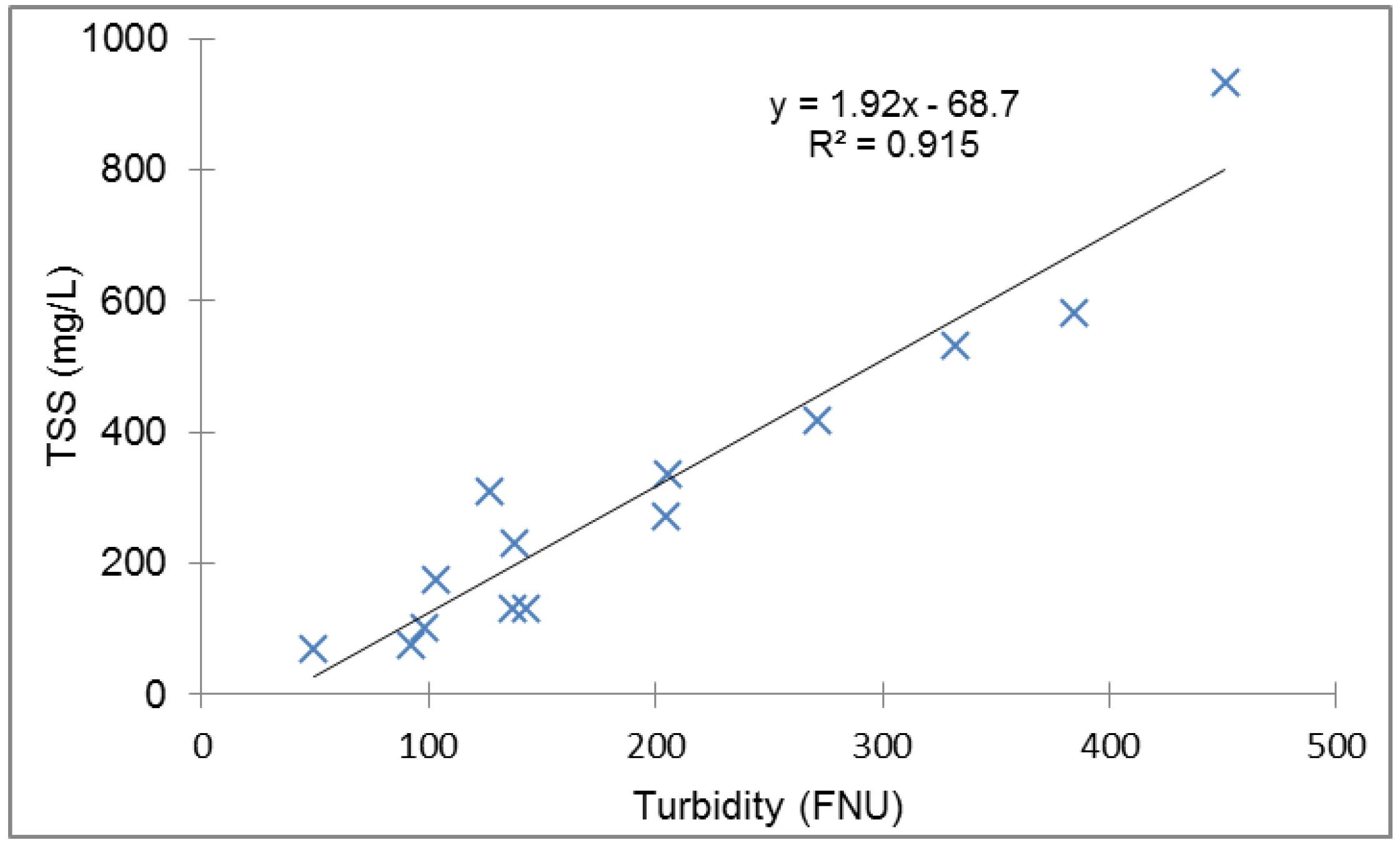
Appendix C. TSS-Turbidity Relationship

Appendix D. Distributions of species in composite cores







Appendix E. All Mass Balance Terms
| Pollutant | Min (g) | Mout,drain (g) | Mout,over (g) | Msoil,initial (g) | Msoil,final (g) |
|---|---|---|---|---|---|
| TSS | 43,200 (36,800, 57,600) | 5500 (4540, 6900) | 8420 (5180, 18,800) | - | - |
| OC | 7320 (6320, 8970) | 1220 (1900, 1620) | 1080 (2820, 4200) | 86,700 (75,100, 100,000) | 52,200 (46,600, 58,700) |
| Cu | 32.5 (28.2, 39.8) | 4.53 (3.51, 6.41) | 7.40 (4.96, 12.7) | 126 (110, 143) | 151 (138, 164) |
| Zn | 90.0 (76.1, 118) | 11.4 (7.99, 21.0) | 19.1 (11.8, 40.2) | 313 (271, 362) | 432 (394, 471) |
| Pyr | 0.125 (0.106, 0.162) | 0.00838 (0.0060, 0.0155) | 0.0256 (0.0157, 0.0536) | 1.45 (1.09, 1.85) | 1.11 (0.830, 1.48) |
| Phen | 0.0439 (0.0366, 0.0586) | 0.00295 (0.00218, 0.00464) | 0.00992 (0.00650, 0.0211) | 0.718 (0.541, 0.920) | 0.824 (0.610, 1.12) |
| BPA | 0.0797 (0.0693, 0.0934) | 0.0208 (0.0180, 0.0252) | 0.0230 (0.0172, 0.0322) | 0.0525 (0.0180, 0.0893) | 0.349 (0.267, 0.503) |
| OP | 0.0821 (0.0697, 0.0983) | 0.0168 (0.0118, 0.0267) | 0.0214 (0.0149, 0.0322) | 0.0333 (0.0117, 0.0562) | 0.241 (0.141, 0.411) |
| NP | 0.301 (0.257, 0.360) | 0.106 (0.0766, 0.168) | 0.0846 (0.0599, 0.125) | 1.58 (1.04, 2.16) | 3.08 (2.31, 4.06) |
| DEHP | 2.95 (2.31, 4.17) | 1.50 (1.15, 2.08) | 0.606 (0.357, 1.50) | 5.56 (2.98, 8.34) | 26.4 (15.1, 44.3) |
| Pollutant | Mintercepted (g) | ΔMsoil (g) |
|---|---|---|
| TSS | 29,000 (24,900, 35,300) | - |
| OC | 4200 (3430, 5060) | −34,500 (−46,500, −23,700) |
| Cu | 20.5 (17.2, 24.1) | 24.8 (10.3, 39.2) |
| Zn | 58.8 (46.6, 72.0) | 119 (73.5, 162) |
| Pyr | 0.0901 (0.0755, 0.108) | −0.337 (−0.824, 0.160) |
| Phen | 0.0307 (0.0255, 0.0377) | 0.107 (−0.180, 0.437) |
| BPA | 0.0355 (0.0277, 0.0442) | 0.297 (0.207, 0.452) |
| OP | 0.0434 (0.0308, 0.0549) | 0.208 (0.106, (0.377) |
| NP | 0.108 (0.0422, 0.156) | 1.51 (0.589, 2.60) |
| DEHP | 0.806 (0.063, 1.58) | 20.9 (9.01, 38.9) |
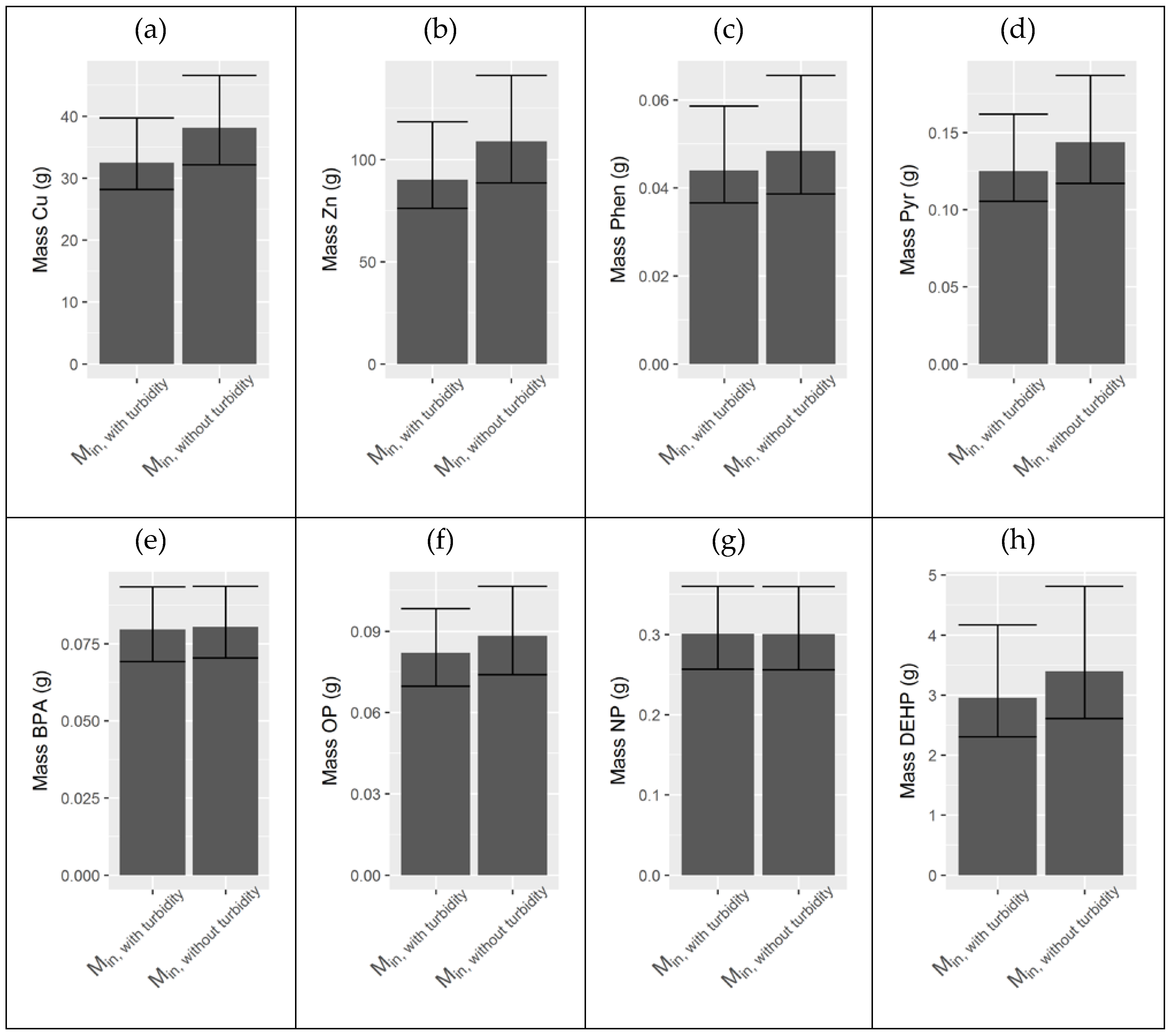
Appendix F. Comparison of Inlet Masses with and without Turbidity Data

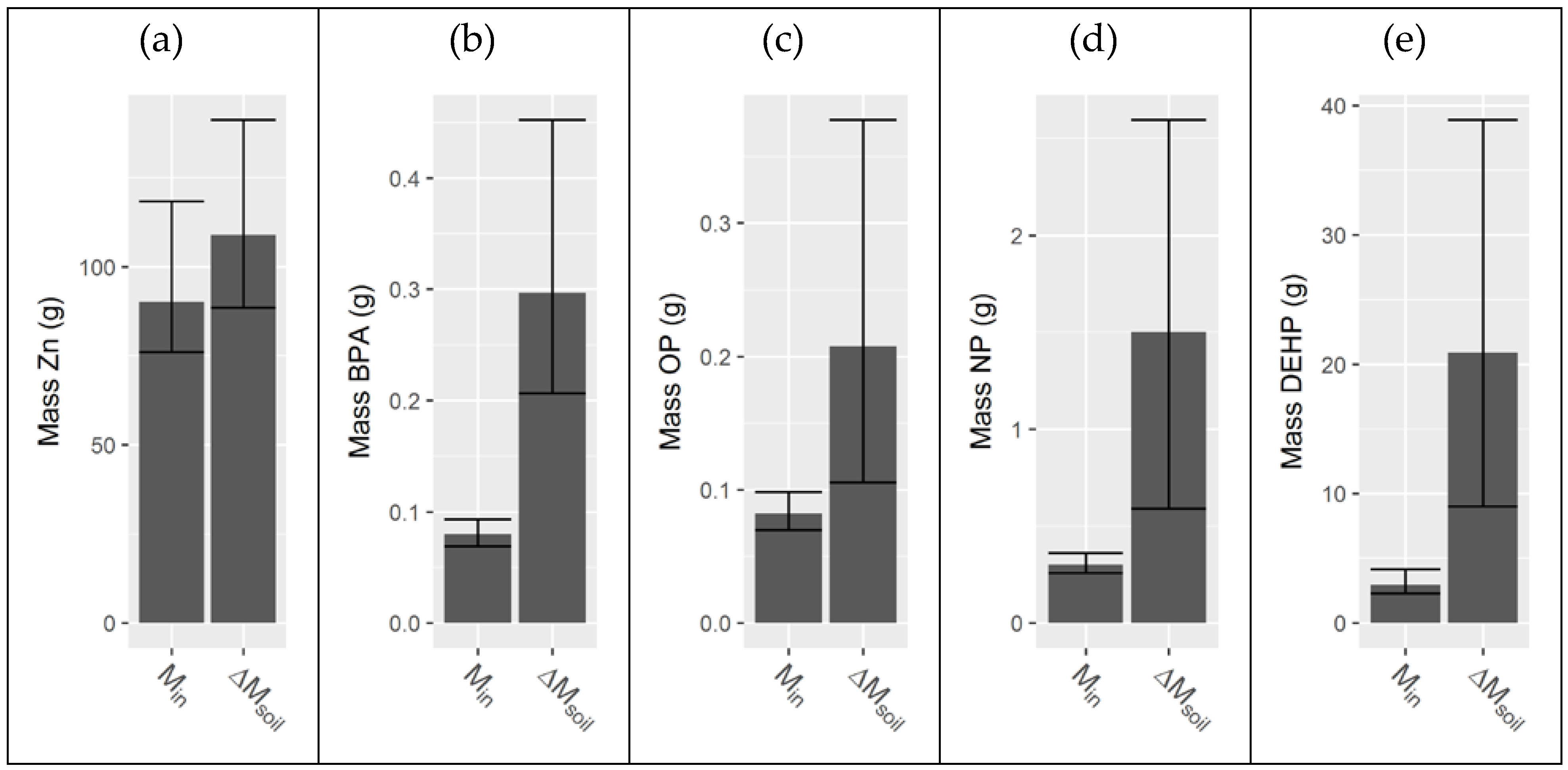
Appendix G. Comparison on Inlet Mass to Mass Accumulated in the Soil

Appendix H. Contamination of Atmospheric Deposition
| Road Runoff | Atmospheric Deposition 1 | |
|---|---|---|
| Cu (µg/L) | 258 (98.1, 547) | 3.9 (1.9, 22) |
| Zn (µg/L) | 693 (236, 1650) | 38 (18, 105) |
| Phen (µg/L) | 0.356 (0.102, 0.594) | 0.014 (<0.01, 0.036) |
| Pyr (µg/L) | 0.851 (0.205, 2.30) | 0.0085 (0.005, 0.035) |
Appendix I. Galvanized Steel Barrier along the Biofiltration Swale

Appendix J. Proportion of Intercepted Pollutant Mass Dissipated in the System (Esoil) and Relative Uncertainty Values
| Pollutant | Esoil (%) |
|---|---|
| Pyr | 474 (−76, 1039) |
| Phen | −242 (−1350, 682) |
| BPA | −739 (−1270, −447) |
| OP | −383 (−852, −133) |
| NP | −1310 (−3610, −410) |
| DEHP | −2520 (−13300, −789) |
Appendix K. Relative Uncertainty Values
| Msoil,initial | Msoil,final | ΔMsoil | Min | Mout,drain | Mout,over | Mintercepted | Esoil | |
|---|---|---|---|---|---|---|---|---|
| Cu | [−12,14] | [−8,9] | [−14,11] | [−13,22] | [−23,41] | [−33,71] | [−16,18] | - |
| Zn | [−14,16] | [−9,9] | [−38,37] | [−15,32] | [−30,85] | [−38,110] | [−21,22] | - |
| Pyr | [−25,27] | [−25,33] | [−145,147] | [−16,30] | [−29,85] | [−39,113] | [−16,20] | [−116,119] |
| Phen | [−25,28] | [−26,35] | [−269,310] | [−17,33] | [−26,57] | [−34,113] | [−17,23] | [−457,382] |
| BPA | [−66,70] | [−23,44] | [−30,52] | [−13,17] | [−13,21] | [−25,40] | [−22,25] | [−72,40] |
| OP | [−65,69] | [−41,71] | [−49,82] | [−15,20] | [−29,59] | [−30,51] | [−29,26] | [−123,65] |
| NP | [−34,37] | [−25,32] | [−61,73] | [−15,20] | [−28,58] | [−29,47] | [−61,44] | [−175,69] |
| DEHP | [−46,50] | [−43,68] | [−57,86] | [−22,41] | [−36,39] | [−41,148] | [−92,95] | [−427,69] |
References
- Fletcher, T.D.; Shuster, W.; Hunt, W.F.; Ashley, R.; Butler, D.; Arthur, S.; Trowsdale, S.; Barraud, S.; Semadeni-Davies, A.; Bertrand-Krajewski, J.-L.; et al. SUDS, LID, BMPs, WSUD and more—The evolution and application of terminology surrounding urban drainage. Urban Water J. 2014, 12, 525–542. [Google Scholar] [CrossRef]
- EC Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the Field of Water Policy 2013. Available online: https://eur-lex.europa.eu/%20LexUriServ/LexUriServ.do?uri=OJ:L:2013:226:0001:0017:EN:PDF (accessed on 30 March 2018).
- Davis, A.P.; Hunt, W.F.; Traver, R.G.; Clar, M. Bioretention technology: Overview of current practice and future needs. J. Environ. Eng. 2009, 135, 109–117. [Google Scholar] [CrossRef]
- Roy-Poirier, A.; Champagne, P.; Filion, Y. Review of bioretention system research and design: Past, present, and future. J. Environ. Eng. 2010, 136, 878–889. [Google Scholar] [CrossRef]
- LeFevre, G.; Paus, K.H.; Natarajan, P.; Gulliver, J.S.; Novak, P.J.; Hozalski, R.M. Review of Dissolved Pollutants in Urban Storm Water and Their Removal and Fate in Bioretention Cells. J. Environ. Eng. 2014, 141. [Google Scholar] [CrossRef]
- Davis, A.P. Field Performance of Bioretention: Water Quality. Environ. Eng. Sci. 2007, 24, 1048–1064. [Google Scholar] [CrossRef]
- DiBlasi, C.J.; Li, H.; Davis, A.P.; Ghosh, U. Removal and Fate of Polycyclic Aromatic Hydrocarbon Pollutants in an Urban Stormwater Bioretention Facility. Environ. Sci. Technol. 2009, 43, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Dietz, M.E.; Clausen, J.C. A field evaluation of rain garden flow and pollutant treatment. Water. Air. Soil Pollut. 2005, 167, 123–138. [Google Scholar] [CrossRef]
- Hatt, B.; Fletcher, T.D.; Deletic, A. Hydrologic and pollutant removal performance of stormwater biofiltration systems at the field scale. J. Hydrol. 2009, 365, 310–321. [Google Scholar] [CrossRef]
- Hunt, W.F.; Jarrett, A.R.; Smith, J.T.; Sharkey, L.J. Evaluating Bioretention Hydrology and Nutrient Removal at Three Field Sites in North Carolina. J. Irrig. Drain. Eng. 2006, 132, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Monrabal-Martinez, C.; Meyn, T.; Muthanna, T.M. Characterization and temporal variation of urban runoff in a cold climate—Design implications for SuDS. Urban Water J. 2018, 1–9. [Google Scholar] [CrossRef]
- Flanagan, K.; Branchu, P.; Boudahmane, L.; Caupos, E.; Demare, D.; Deshayes, S.; Dubois, P.; Meffray, L.; Partibane, C.; Saad, M.; et al. Retention and transport processes of particulate and dissolved micropollutants in stormwater biofilters treating road runoff. Sci. Total Environ. 2019, 656, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Tedoldi, D.; Chebbo, G.; Pierlot, D.; Kovacs, Y.; Gromaire, M.-C. Impact of runoff infiltration on contaminant accumulation and transport in the soil/filter media of Sustainable Urban Drainage Systems: A literature review. Sci. Total Environ. 2016, 569, 904–926. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.; Seagren, E.A.; Davis, A.P. Sustainable Oil and Grease Removal from Synthetic Stormwater Runoff Using Bench-Scale Bioretention Studies. Water Environ. Res. 2006, 78, 141–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeFevre, G.H.; Novak, P.J.; Hozalski, R.M. Fate of Naphthalene in Laboratory-Scale Bioretention Cells: Implications for Sustainable Stormwater Management. Environ. Sci. Technol. 2012, 46, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Leroy, M.C.; Legras, M.; Marcotte, S.; Moncond’huy, V.; Machour, N.; Le Derf, F.; Portet-Koltalo, F. Assessment of PAH dissipation processes in large-scale outdoor mesocosms simulating vegetated road-side swales. Sci. Total Environ. 2015, 520, 146–153. [Google Scholar] [CrossRef] [PubMed]
- LeFevre, G.H.; Hozalski, R.M.; Novak, P.J. The role of biodegradation in limiting the accumulation of petroleum hydrocarbons in raingarden soils. Water Res. 2012, 46, 6753–6762. [Google Scholar] [CrossRef] [PubMed]
- Maniquiz, M.C.; Kim, L.-H.; Lee, S.; Choi, J. Flow and mass balance analysis of eco-bio infiltration system. Front. Environ. Sci. Eng. 2012, 6, 612–619. [Google Scholar] [CrossRef]
- Johnston, C.A. Development and Evaluation of Infilling Methods for Missing Hydrologic and Chemical Watershed Monitoring Data. Master’s Thesis, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA, 1999. [Google Scholar]
- Sun, S.; Barraud, S.; Castebrunet, H.; Aubin, J.-B.; Marmonier, P. Long-term stormwater quantity and quality analysis using continuous measurements in a French urban catchment. Water Res. 2015, 85, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bertrand-Krajewski, J.-L.; Bardin, J.-P.; Mourad, M.; Béranger, Y. Accounting for sensor calibration, concentration heterogeneity, measurement and sampling uncertainties in monitoring urban drainage systems. Water Sci. Technol. 2003, 47, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Versini, P.-A.; Joannis, C.; Chebbo, G. Guide technique sur la mesurage de la turbidité dans les réseaux d’assainissement; Onema: Vincennes, France, 2015. [Google Scholar]
- Flanagan, K.; Branchu, P.; Boudahmane, L.; Caupos, E.; Demare, D.; Deshayes, S.; Dubois, P.; Meffray, L.; Partibane, C.; Saad, M.; et al. Field performance of two biofiltration systems treating micropollutants from road runoff. Water Res. 2018, 145, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Tedoldi, D.; Chebbo, G.; Pierlot, D.; Kovacs, Y.; Gromaire, M.-C. Assessment of metal and PAH profiles in SUDS soil based on an improved experimental procedure. J. Environ. Manag. 2017, 202, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sage, J. Concevoir et optimiser la gestion hydrologique du ruissellement pour une maîtrise à la source de la contamination des eaux pluviales urbaines. Ph.D. Thesis, Université de Paris-Est, Champs-sur-Marne, France, 2016. [Google Scholar]
- Hollis, G.E.; Ovenden, J.C. One year irrigation experiment to assess losses and runoff volume relationships for a residential road in hertfordshire, England. Hydrol. Process. 1988, 2, 61–74. [Google Scholar] [CrossRef]
- Ramier, D.; Berthier, E.; Andrieu, H. The hydrological behaviour of urban streets: Long-term observations and modelling of runoff losses and rainfall-runoff transformation. Hydrol. Process. 2011, 25, 2161–2178. [Google Scholar] [CrossRef]
- Hannouche, A.; Chebbo, G.; Joannis, C.; Gasperi, J.; Gromaire, M.-C.; Moilleron, R.; Barraud, S.; Ruban, V. Stochastic evaluation of annual micropollutant loads and their uncertainties in separate storm sewers. Environ. Sci. Pollut. Res. 2017, 24, 28205–28219. [Google Scholar] [CrossRef] [PubMed]
- Van Buren, M.A.; Watt, W.E.; Marsalek, J. Application of the log-normal and normal distributions to stormwater quality parameters. Water Res. 1997, 31, 95–104. [Google Scholar] [CrossRef]
- Bertrand-Krajewski, J.-L.; Chebbo, G.; Saget, A. Distribution of pollutant mass vs volume in stormwater discharges and the first flush phenomenon. Water Res. 1998, 32, 2341–2356. [Google Scholar] [CrossRef]
- Huber, M.; Helmreich, B. Stormwater Management: Calculation of Traffic Area Runoff Loads and Traffic Related Emissions. Water 2016, 8, 294. [Google Scholar] [CrossRef]
- Björklund, K.; Cousins, A.P.; Strömvall, A.-M.; Malmqvist, P.-A. Phthalates and nonylphenols in urban runoff: Occurrence, distribution and area emission factors. Sci. Total Environ. 2009, 407, 4665–4672. [Google Scholar] [CrossRef] [PubMed]
- Bressy, A.; Gromaire, M.-C.; Lorgeoux, C.; Saad, M.; Leroy, F.; Chebbo, G. Towards the determination of an optimal scale for stormwater quality management: Micropollutants in a small residential catchment. Water Res. 2012, 46, 6799–6810. [Google Scholar] [CrossRef]
- Lamprea, K.; Bressy, A.; Mirande-Bret, C.; Caupos, E.; Gromaire, M.-C. Alkylphenol and bisphenol A contamination of urban runoff: An evaluation of the emission potentials of various construction materials and automotive supplies. Environ. Sci. Pollut. Res. 2018, 25, 21887–21900. [Google Scholar] [CrossRef]
- Hatt, B.; Morison, P.; Fletcher, T.; Deletic, A. Stormwater Biofiltration Systems: Adoption Guidelines; Monash University: Clayton, Victoria, Australia, 2009. [Google Scholar]
- Thomas, E.; Ratovelomanana, T. RD 212: Expérimentation d’un Assainissement Alternatif Routier; CD 77: Vert-Saint-Denis, France, 2016. [Google Scholar]
- Bergé, A.; Cladière, M.; Gasperi, J.; Coursimault, A.; Tassin, B.; Moilleron, R. Meta-analysis of environmental contamination by alkylphenols. Environ. Sci. Pollut. Res. 2012, 19, 3798–3819. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz, A.; Björklund, K.; Eriksson, E.; Kalmykova, Y.; Strömvall, A.-M.; Siopi, A. Emissions of organic pollutants from traffic and roads: Priority pollutants selection and substance flow analysis. Sci. Total Environ. 2017, 580, 1162–1174. [Google Scholar] [CrossRef]
- Taylor, J.R. An Introduction to Error Analysis, 2nd ed.; University Science Books: Sausalito, CA, USA, 1997. [Google Scholar]
- Gaspéri, J.; Ayrault, S.; Moreau-Guigon, E.; Alliot, F.; Labadie, P.; Budzinski, H.; Blanchard, M.; Muresan, B.; Caupos, E.; Cladière, M.; et al. Contamination of soils by metals and organic micropollutants: Case study of the Parisian conurbation. Environ. Sci. Pollut. Res. 2016. [Google Scholar] [CrossRef]
- Mackay, D. (Ed.) Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals, 2nd ed.; CRC/Taylor & Francis: Boca Raton, FL, USA, 2006; ISBN 978-1-56670-687-2. [Google Scholar]









| Properties | Biofiltration Swale |
|---|---|
| Filter media texture | Sandy loam |
| Filter media pH in water | 8.73 |
| Initial organic carbon content (by mass, %) | 0.9 |
| Hydraulic conductivity (mm/h) | 21 |
| Filter media depth (cm) | 50 |
| Ponding depth (cm) | 17 |
| Width (perpendicular to road, m) | 0.5 |
| Length (parallel to road, m) | 32 |
| Surface area ratio (Adevice/Acatchment, %) | 4.5 |
| Parameter | Substances and Abbreviations | Method | Limit of Quantification (LoQ) | Uncertainty |
|---|---|---|---|---|
| Suspended solids | Total suspended solids (TSS) | Filter: 0.7 µm fiberglass Method: Filtration Standard: NF EN 872 | 2 mg/L | |
| Organic carbon | Organic carbon (OC) | Filter: 0.7 µm fiberglass Method: Thermal combustion-Infrared detector Standard: NF EN 1484 (dissolved) | Dissolved: 0.3 mg/L Particulate: mg/g | ±10% |
| Trace metals | Copper (Cu), Zinc (Zn) | Filter: 0.45 µm cellulose acetate Methods: ICP-MS and ICP-OES Extraction: Total solubilization by HF and HClO4 acid digestion, evaporation and resuspension with HNO3 (particulate) Standards: NF X31-147 (acid digestion), NF EN ISO 17294 (ICP-MS), NF EN ISO 11885 (ICP-OES) | Dissolved: 0.2 (Cu), 0.3 (Zn) µg/L Particulate: 0.4 (Cu), 1 (Zn) mg/kg | Dissolved: ±9 (Cu), 14 (Zn) % Particulate: ± 14 (Cu), 13 (Zn) % |
| PAHs | Phenanthrene (Phen), Pyrene (Pyr) | Filter: 0.7 µm fiberglass Extraction: Liquid-liquid (dissolved), ultrasound solid-liquid (particulate) Method: GC-MSStandard: XP ISO-TS 28581 and NF ISO 28540 (dissolved), XP X33-012 and NF EN 15527 (particulate) | Dissolved: 10 ng/L Particulate: µg/g | SL: 20 ng/L (dissolved), 0.03 µg/g (particulate) Absolute uncertainty between LoQ and SL: ± 6 ng/L (dissolved), 0.006 µg/g (particulate) Relative uncertainty beyond SL: ± 40 (dissolved), 20 (particulate) % |
| BPA/AP | Bisphenol-A (BPA), Para-nonylphenol (NP), 4-tert-octylphenol (OP) | Filter: 0.7 µm fiberglass Extraction: Solid-phase (dissolved), Microwave (particulate) Method: UPLC-MSMS | Dissolved: 11 (BPA), 79 (NP), 7 (OP) ng/L Particulate: (BPA), (NP), (OP) µg/g | Dissolved: ± 28 (BPA), 37 (NP), 61 (OP) % Particulate: ± 25 (BPA), 44 (NP), 48 (OP) % |
| Phthalates | Bis(2-ethylhexyl) phthalate (DEHP) | Filter: 0.7 µm fiberglass Extraction: Solid-phase (dissolved), Microwave (particulate) Method: GC-MS | Dissolved: 350 (DEHP) ng/L Particulate: (DEHP) µg/g | Dissolved: ± 53 (DEHP) % Particulate: ± 35 (DEHP) % |
| Pollutant | Min over Full Period (g) | Min (g/ha/year) | Min (mg/ act.ha/mm) |
|---|---|---|---|
| TSS | 4.3 × 104 (3.7 × 104, 5.8 × 104) | 1.1 × 106 (9.6 × 105, 1.5 × 106) | 2.7 × 106 (2.3 × 106, 3.5 × 106) |
| OC | 7.3 × 103 (6.3 × 103, 9.0 × 103) | 1.9 × 105 (1.7 × 105, 2.4 × 105) | 4.5 × 105 (4.0 × 105, 5.5 × 105) |
| Cu | 32.5 (28.2, 39.8) | 851 (738, 1040) | 2010 (1740, 2450) |
| Zn | 90.1 (76.1, 118) | 2360 (1990, 3100) | 5560 (4770, 7300) |
| Pyr | 0.125 (0.106, 0.162) | 3.27 (2.76, 4.24) | 7.71 (6.51, 10.0) |
| Phen | 0.0439 (0.0366, 0.0586) | 1.15 (0.96, 1.53) | 2.71 (2.26, 3.61) |
| BPA | 0.0797 (0.0693, 0.934) | 2.09 (1.81, 2.45) | 4.92 (4.27, 5.76) |
| OP | 0.0821 (0.0697, 0.0983) | 2.15 (1.83, 2.57) | 5.06 (4.30, 6.07) |
| NP | 0.300 (0.257, 0.360) | 7.87 (6.73, 9.43) | 18.6 (15.9, 22.2) |
| DEHP | 2.95 (2.31, 4.17) | 77.3 (60.4, 109.3) | 182 (142, 257) |
| Parameter | Eint (%) | Mout,over/Min (%) | Mout,drain/Min (%) | Ec (%) 1 |
|---|---|---|---|---|
| Volume | 21 (15, 24) | 35 (31, 37) | 45 (41, 52) | - |
| TSS | 67 (56, 74) | 20 (13, 34) | 13 (9, 16) | 92 (27, 95) |
| OC | 57 (48, 64) | 22 (16, 32) | 20 (15, 27) | 70 (25, 77) |
| Cu | 63 (54, 69) | 23 (17, 33) | 14 (10, 20) | 76 (29, 92) |
| Zn | 65 (51, 73) | 21 (15, 34) | 13 (8, 24) | 89 (35, 97) |
| Pyr | 72 (60, 79) | 21 (14, 34) | 7 (4, 13) | 94 (49, 95) |
| Phen | 70 (58, 77) | 23 (16, 37) | 7 (4, 11) | 92 (43, 95) |
| BPA | 45 (38, 51) | 29 (23, 36) | 26 (21, 33) | 57 (−44, 77) |
| OP | 53 (40, 62) | 26 (20, 34) | 20 (14, 33) | 76 (−77, 93) |
| NP | 36 (15, 48) | 28 (22, 36) | 35 (24, 57) | 56 (−214, 71) |
| DEHP | 27 (3, 44) | 21 (13, 38) | 51 (33, 77) | 9 (−128, 32) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flanagan, K.; Branchu, P.; Boudahmane, L.; Caupos, E.; Demare, D.; Deshayes, S.; Dubois, P.; Kajeiou, M.; Meffray, L.; Partibane, C.; et al. Stochastic Method for Evaluating Removal, Fate and Associated Uncertainties of Micropollutants in a Stormwater Biofilter at an Annual Scale. Water 2019, 11, 487. https://doi.org/10.3390/w11030487
Flanagan K, Branchu P, Boudahmane L, Caupos E, Demare D, Deshayes S, Dubois P, Kajeiou M, Meffray L, Partibane C, et al. Stochastic Method for Evaluating Removal, Fate and Associated Uncertainties of Micropollutants in a Stormwater Biofilter at an Annual Scale. Water. 2019; 11(3):487. https://doi.org/10.3390/w11030487
Chicago/Turabian StyleFlanagan, Kelsey, Philippe Branchu, Lila Boudahmane, Emilie Caupos, Dominique Demare, Steven Deshayes, Philippe Dubois, Meriem Kajeiou, Laurent Meffray, Chandirane Partibane, and et al. 2019. "Stochastic Method for Evaluating Removal, Fate and Associated Uncertainties of Micropollutants in a Stormwater Biofilter at an Annual Scale" Water 11, no. 3: 487. https://doi.org/10.3390/w11030487